Applying for provisional registration for a prescription medicine
Guidance on provisional registration process for prescription medicines with provisional determination.
Purpose
This guidance assists sponsors with the provisional registration process. It outlines the key differences between this process and the standard registration process for prescription medicines.
You must submit an application for provisional determination and have the determination approved before you lodge a submission for provisional registration. See Provisional determination: a step-by-step guide for prescription medicines for more information.
This guidance should be read in conjunction with guidance on the standard prescription medicines registration process.
Prescription medicines registration process
The standard registration process for prescription medicines consists of eight phases with eight milestones.
Internal business practices (milestone process) for the standard registration process aim to process submissions within a target timeframe of 220 working days. The definition of a working day is described in regulation 16A of the Therapeutic Goods Regulations 1990 (the Regulations). The timeframe is calculated from acceptance for evaluation through to the delegate's decision.
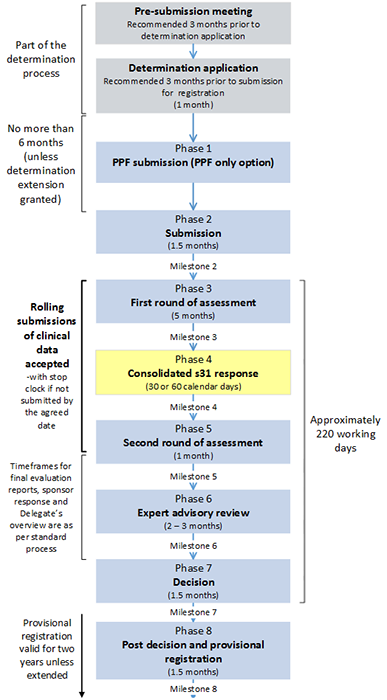
Provisional registration process diagram
Note: the timeframes below represent maximum timeframes for each phase of the process as a guideline only.
Figure 1. Provisional registration process
{kind=link}
This text representation of this diagram is provided as a list with numbered steps.
- Pre-submission meeting [Part of the determination process]
- Recommended 3 months prior to determination application
- Determination application (1 month) [Part of the determination process]
- Recommended 3 months prior to submission for registration
- Phase 1: PPF submission (PPF only option)
[No more than 6 months between end of Determination application and beginning of Phase 2(unless determination extension granted]
- Phase 2: Submission (1.5 months)
- Milestone 2
- Phase 3: First round of assessment (5 months)
- Milestone 3
- Phase 4: Consolidated s31 response (30 or 60 calendar days)
- Milestone 4
- Phase 5: Second round of assessment (1 month)
- Milestone 5
[Beginning of Phase 3 to end of Phase 5: Rolling submissions of clinical data accepted – with stop clock if not submitted by the agreed date]
- Phase 6: Expert advisory review (2-3 months)
- Milestone 6
[Phase 5 and Phase 6: Timeframes for final evaluation reports, sponsor response and Delegate's overview are as per standard process]
- Phase 7: Decision (1.5 months)
- Milestone 7
[Approximately 220 working days between beginning of Phase 3 and end of Phase 7]
- Phase 8: Post decision and provisional registration (1.5 months)
- Milestone 8.
[Provisional registration valid for two years unless extended].
Provisional registration process
As part of the provisional approval pathway, the provisional registration process will allow certain medicines to be provisionally registered in the Australian Register of Therapeutic Goods (ARTG) for a limited duration. These medicines are registered on the basis of preliminary clinical data, where the benefit of early availability of the medicine outweighs the risk inherent in the fact that additional data are still required.
See Provisional determination eligibility criteria and supporting documentation guidance for further information on preliminary clinical data.
Similar to the standard prescription medicines registration process, the provisional registration process has eight phases but with some changes to accommodate the uncertainty associated with preliminary clinical data (see the Provisional registration process diagram).
These changes include:
- allowing a limited number of rolling submissions of clinical data during the evaluation phases, if this is agreed upfront between you and TGA
- closer coordination between clinical, nonclinical, quality and Risk Management Plan (RMP) evaluation areas will ensure that any emerging safety signals are identified and managed (to assist with managing the risks of preliminary clinical data)
- the delegate may seek external expert advice on issues concerning the submission earlier and more frequently in the pre-market registration process as required
- the delegate may also refer submissions to a scheduled meeting of the Advisory Committee on Medicines (ACM) or Advisory Committee on Vaccines (ACV) for advice during the first or second rounds of assessment as required
Timeframes for the provisional registration process
Although the clinical data provided as part of a submission for provisional registration may be preliminary, this does not mean that the evaluation workload would be less than for a standard submission for registration. Despite being based on preliminary data, a higher level of scrutiny and deliberation may be required to understand uncertainties and weigh up the benefits and risks of earlier availability of these medicines, resulting in a similar evaluation time.
Given that full quality and nonclinical data (Modules 3 and 4) will be required, evaluation workload of these parts of the dossier would be similar to standard submissions for registration.
The provisional registration process is also designed with a target timeframe of 220 working days from acceptance for evaluation through to the delegate's decision. The statutory timeframe for both provisional registration and standard registration is 255 working days (regulation 16C(3)).
To facilitate earlier access to vital medicines, provisional registration applications will be prioritised within the target timeframe.
Eligibility for provisional registration
If you apply for registration of a medicine under section 23, and a provisional determination is in force relating to you, the medicine and the indication mentioned in the application, then the application is taken to be an application for provisional registration of that medicine (section 23AA of the Therapeutic Goods Act 1989 (the Act)).
This ties the application for registration to the specifications of the provisional determination which was granted previously under subsection 22D of the Act.
Provisional determination must be in force
To use the provisional pathway, you must ensure that a provisional determination is in force for the relevant medicine, sponsor and indication at the time of making your section 23 registration application. The registration application is 'made' when it is submitted using the approved form or manner and is accompanied by specified information (the dossier).
The determination will cease to be in force six months from the date we notify you of our decision to make the provisional determination, unless it is revoked or extended, or a section 23 application is made.
If you wish to use the provisional registration process and your provisional determination is no longer in force (and the time to apply for an extension has expired), you must submit a new determination application and pay the determination fee.
Please see the Provisional determination process guidance for more information on how to apply for determination and extensions of determination.
Data requirements for provisional registration
Preliminary clinical data for provisional registration
For a provisional registration application, you must satisfactorily establish the safety and efficacy of the medicine, including a positive risk/benefit balance based on preliminary clinical data.
To establish safety and efficacy, the preliminary clinical evidence provided in support of the provisional registration application needs to be sufficient to allow the benefits of the product to be assessed against the risks identified by the evidence. We will re-assess risks related to the absence of evidence through data provided at a later stage, as part of the confirmatory data. Confirmatory data should confirm the relationship between outcomes predicted by the surrogate endpoint, or other preliminary data, and the clinical benefit as demonstrated by direct clinical outcomes.
In addition to other requirements (see Australian Regulatory Guidelines for Prescription medicines (ARGPM)) you must support the provisional registration application with the following clinical information:
- clinical study reports for all relevant studies
- the clinical overview (Module 2.5) should include a discussion of the efficacy, safety profile and risk/benefit profile, taking into account the uncertainty and the strength of the available evidence. Data that is not available at the time of provisional registration should be discussed. The acceptability of less comprehensive data should be justified
- a clinical study plan as part of the RMP to support management of the additional risk introduced by the lack of comprehensive data. The plan should address the uncertainties in the preliminary data.
Note that the use of a surrogate as a primary endpoint in a pivotal study of clinical efficacy/safety will not automatically require a registration application to be submitted for provisional approval.
Rolling submissions of clinical data
For provisional registration, it is likely that the dossier will consist of a combination of completed and ongoing pivotal or supporting clinical trials which may be in progress during the pre-market registration period.
On a case-by-case basis, we may agree upfront to accept a limited number of rolling submissions of clinical data during the evaluation period, where this information might have a material impact on the registration decision.
To assist TGA evaluation planning and resourcing, we will need to prospectively agree to additional data that will be generated after submission of the dossier and the proposed timeframe for any rolling submissions of data. This should be:
- raised at your pre-submission meeting
- included in your determination application
- updated when you lodge the dossier.
We will not accept rolling clinical data that has not been agreed to prior to dossier acceptance during the provisional registration process, unless requested by the evaluator or related to the safety of the medicine. We also will not accept data with a larger scope than what was agreed upfront.
If you submit incomplete, delayed, or poor quality applications you cannot provide unsolicited information during the registration process to rectify these deficiencies. Submission of information that has not been agreed upfront will be treated as unsolicited data. We will accept unsolicited information only under very specific circumstances (for example related to safety) as set out in the standard prescription medicines registration process 2.5.2 Unsolicited Information.
You must notify us as soon as possible of any changes to the rolling data that was agreed upfront, for example, the intended dates for data submission, or any other change to the agreed content. Any delay in submission of data may result in a stop-clock.
Quality and nonclinical data
You must include comprehensive quality and nonclinical safety modules (Module 3 and Module 4) in your provisional registration application that fulfil our mandatory data requirements and scientific guidelines for the specific application type adopted by us from the European Union and the International Conference on Harmonisation.
Where you choose not to provide data according to a particular guideline or aspect of a guideline, you must provide a scientifically robust justification to explain why the guideline is not applicable and why the proposed alternative is applicable.
Good Manufacturing Practice requirements
The Good Manufacturing Practice (GMP) requirements at the time of lodging a submission for registration via the provisional registration pathway are the same as those for the standard prescription medicines registration process and the Priority review registration process.
At the time of lodging a submission for provisional registration you will need to:
- provide the currently issued Good Manufacturing Practice (GMP) clearance or licence tracking number for all manufacturing sites relevant to the provisional registration submission; OR
- demonstrate that you have applied for a GMP clearance, licence or certification by providing the tracking numbers to obtain GMP approval for all manufacturing sites. We will verify that your GMP application is valid as part of the submission for registration acceptance process (i.e. confirm that you have paid the required GMP application fees and all of the required evidence to support the GMP application for all relevant manufacturing sites has been lodged in support of the application)
See Licencing and Certification guidance and GMP clearance guidance for the evidence required when lodging your application.
Overseas manufacturing sites
If the manufacturing site/s are overseas and you are aware that they will be inspected soon (i.e. within the provisional registration timeframe) by a Mutual Recognition Agreement (MRA) partner regulator as part of a parallel submission for registration overseas, you can submit a GMP clearance application and request that the TGA liaise with the overseas regulator.
Alternatively, if:
- a recognised regulator has not inspected and is not planning to inspect the overseas manufacturing site OR
- the GMP certificate or inspection report will not be available soon and you are unable to provide the necessary evidence as part of a GMP Clearance application via the MRA or Compliance Verification Pathway (CV), then you may request that TGA perform an on-site inspection via a GMP Certification Application.
Use of reports from comparable overseas regulators
We encourage you to submit reports from acceptable overseas regulators to supplement your provisional submission for registration. This includes where the medicine has been conditionally registered overseas. Reports from overseas regulators can be submitted during the provisional registration process and also after the medicine is provisionally registered to support our ongoing post-market monitoring.
Applications to the provisional registration pathway for new chemical entities and new biological entities are unlikely to be eligible for our comparable overseas report (COR)-based evaluation process for prescription medicines. This is because the COR-based evaluation process requires full marketing approval (i.e. not provisional or conditional approval) to have been granted by the relevant overseas regulator/s.
Application and evaluation fees
Application and evaluation fees are higher for the provisional registration process than for the standard prescription medicines registration process. See fees and payments for fee amounts (and Schedule 9 of the Regulations).
The processes for invoicing and payment of these fees are the same as in the standard prescription medicines registration process.
Fee waivers for medicines with a valid orphan drug designation apply to the application and evaluation fees for the provisional registration process, provided that the therapeutic indication for provisional registration is identical to, or a subset of, the orphan indication.
Obtaining an orphan drug fee waiver for subsequent submissions during the provisional registration period requires reapplication and reassessment of the orphan drug designation.
Phase 1: Pre-submission Planning Form (PPF)
For provisional registration submissions, this phase differs from the standard prescription medicines registration process in the following ways:
- the PPF for provisional registration will follow the PPF-only pre-submission phase option (the PPF-only option), which requires the submission for registration to be in electronic Common Technical Document (eCTD) format. You will be required to fill in the sections relevant to active provisional determinations when submitting the PPF via the pre-submission form in TGA Business Services
- as in the standard PPF-only process, there will be no formal Milestone 1. You should proceed to lodge your entire submission for registration as soon as the complete submission number is visible on TGA Business Services. This will occur once we have added the relevant stream number based on your proposed indication (i.e. 'PM-yyyy-xxxxx-z-stream number'). Please email us at AET.Application.Entry.Team@health.gov.au if you experience any difficulties
Phase 2: Submission phase
Advise us as soon as possible of any changes to the planned lodgment date for your submission for registration (which you previously provided in your determination application form). Early notification of your planned lodgment date assists us to allocate the required resources.
If you lodge a section 23 submission for registration and a valid provisional determination is in force for the relevant medicine and indication, it will be treated as a submission for provisional registration (section 23AA of the Act).
If the indication changes between provisional determination and dossier lodgement, given that trials are ongoing, we will have the discretion of determining whether the determination is sufficiently applicable to the dossier.
Please note that the indication which is the subject of the submission for provisional registration must be captured within the indication that was the subject of the provisional determination. If the proposed indication is not covered by the determination approved, re-determination will be required.
We will consider acceptable indications as part of accepting the submission for evaluation. The indication that is ultimately provisionally registered may also differ as a result of assessment of the quality, safety and efficacy data submitted with the submission for registration.
Only one determination may apply to each submission for registration i.e. multiple applications which have obtained separate provisional determinations cannot be lodged together in a single submission.
Concurrent applications without provisional determination
Lodge related applications that do not have provisional determination (either because they are ineligible or because you did not apply for determination) separately to your provisional submission for registration via the standard prescription medicines registration process or Priority review registration process, as applicable.
Lodgement of your submission for registration
The submission phase is the same as for the standard prescription medicine registration process except for the following:
- your provisional registration submission must be in the eCTD format
- you must include a copy of the 'provisional determination notification letter' and the 'provisional determination extension letter' (if applicable) in Module 1.5.2 of the submission to allow these details to be captured in eCTD
- you must include an updated and more detailed version of your plan to collect confirmatory data after the medicine is provisionally registered, or reconfirm that no details in the previous plans have changed (see Clinical study plan for provisional registration on this page)
- the requirements for clinical data to be submitted with the dossier will be different to a standard submission for registration, and must align with what was agreed at pre-submission and determination. This will be assessed on a case-by-case basis
- you must provide an outline of any additional clinical data which you intend to submit during the evaluation phases of the provisional registration process, including details of when this data will be submitted and the volume of this data. Approximate submission dates may be acceptable (for example where the timing is event-driven), however you must provide us with updates as they become available
- ensure that your submission supports the evidence that was presented in the determination application to demonstrate that you continue to meet the eligibility criteria for provisional determination
As stated above, the requirements for quality and nonclinical data (Modules 3 and 4) are the same for the standard and provisional registration pathways.
Clinical study plan for provisional registration
Lodgement of a RMP and Australian Specific Annex (ASA) is required as part of the registration application. The pharmacovigilance plan should detail the studies being conducted to collect confirmatory efficacy data, as well as any other ongoing studies. The study milestones and anticipated submission dates should be included, and the ASA should detail the expected submission dates in Australia. We will assess if your plan to submit comprehensive clinical data on the safety and efficacy of the medicine during the provisional registration period (starting on the day that provisional registration commences) is adequate under Section 25(1)(d)(iii) of the Act.
After the registration process is complete, the planned submission timeframes will become a part of the agreed ASA. Changes in these dates require submission of a post-approval RMP update.
The key milestones and completion dates for collecting confirmatory data will also be specified as conditions of registration. If you want to make changes to these agreed milestones, you must lodge a request to vary your conditions of registration in addition to submission of the post-approval RMP update.
See Clinical study plan for provisional registration on this page for an example of the types of information that could be provided as an overview clinical trial plan. You must also include the study protocols along with the overview of the clinical trial plan when you lodge your application. You are also required to provide an updated plan following completion of the second round of assessment.
It should be noted that follow-up studies included in the clinical study plan may involve differences in clinical setting, such as line of therapy. This may impact on the scope of the revised indication once the confirmatory data is received.
Accepting the submission for evaluation
When making a decision as to whether a submission should be accepted into the provisional registration pathway, we will consider whether:
- the product has an applicable determination to support a provisional registration application
- data in the dossier supports the evidence and/or undertakings that were presented in the determination application to justify that the eligibility criteria have been fulfilled
- you have fulfilled our data requirements for provisional registration submissions (quality, clinical and nonclinical)
- if relevant, you have reached agreement with us about a rolling submission of clinical data during the evaluation period
- you have provided sufficient information for us to determine whether the necessary confirmatory data to achieve full registration can be collected within the provisional registration period.
If a submission for provisional registration does not meet all of the above criteria, we may not accept it for evaluation.
We will provide you with notification of our acceptance of the submission in a notification letter at Milestone 2 as per the standard prescription medicines registration process.
Decisions regarding whether submissions are accepted into the provisional registration pathway after this preliminary assessment process are appealable by the applicant only under section 60 of the Act.
The evaluation phase will commence after the submission for registration has been received, checked and accepted for assessment. The clock will start once you have been notified that the submission for registration has been accepted for evaluation. Submissions for provisional registration will be subject to 'batch processing' as per the standard prescription medicines registration process.
Phase 3: First round assessment
This phase is the same as for the prescription medicine registration process except for the following:
Rolling clinical data
While we will agree on the dates for acceptance of rolling submissions of clinical data on a case-by-case basis, as a general guide, we will only accept this data up to the end of Milestone 5 (completion of the final evaluation report) in the registration process. We will only accept rolling clinical data submissions as a new eCTD sequence. A 'stop-clock' may be required if the data received requires greater evaluation resources than was anticipated and/or rolling data is not submitted at the anticipated and agreed time(s).
Phase 4: Consolidated section 31 request response
This phase is the same as for the standard prescription medicine registration process.
Phase 5: Second round assessment
This phase is similar to the standard prescription medicine registration process.
If additional questions arise during this phase as the result of evaluating rolling submissions of clinical data, these will be managed via a mutual stop-clock. Timeframes for your response to these questions will be determined on a case-by-case basis.
In line with the standard registration process, at the completion of the second round of assessment, you will receive the final evaluation report that includes consideration of your responses to the section 31 questions, as well as any further questions that are issued during the second round of evaluation. You will have at least two weeks after receipt of the final evaluation report to notify us of any errors in fact or major omissions in the report.
At the end of the second round of assessment, you will need to provide us with updated information on your clinical trials plan which was provided in your initial submission for provisional registration. This must be provided alongside your statement of errors of fact or omission in response to the final evaluation report (or separately, if there is no response to the final evaluation report) and will inform the delegate's overview.
Expert advice
The evaluators and delegate may seek ad-hoc expert advice as required during the first and second rounds of assessment. Advice received will be captured in the final evaluation report and/or the delegate's overview.
This advice is available to delegates via statutory committees and a specialist advisory panel. Expert advice may be sought from other experts, including international regulators, experts in the therapeutic area and/or researchers involved in ongoing clinical trials. Potential conflicts of interest will be managed to ensure this process is robust.
Phase 6: Expert advisory review
This phase is the same as the standard prescription medicine registration process except for the following:
- the date on which your submission will reach this phase is subject to change as a consequence of the duration of the evaluation phase. The duration of the evaluation phase may be affected if the rolling submissions of clinical data are delayed or take longer to evaluate than anticipated. You will be provided with updates to your Evaluation Plan during the registration process as required
- provisional registration allows for flexibility in the expert advisory review phase. In addition to the scheduled ACM or ACV meetings, an out-of-session expert advisory committee meeting may be required in certain circumstances
Regardless of the mechanism for obtaining expert advice, the timeframes and procedures for exchange of information between you and TGA during this phase are the same as for the standard prescription medicines registration process.
You will receive a copy of the delegate's overview of the submission, as in the standard registration process.
Phase 7: Decision
The process for this phase is the same as for the standard prescription medicine registration process. However, the considerations leading us to a decision to provisionally register a medicine are different from the considerations taken into account when granting a standard registration.
What we will consider when provisionally registering a medicine
In addition to the standard requirements for registration of a prescription medicine, the delegate will base their decision to grant time-limited provisional registration of a medicine on their assessment of whether:
- based on preliminary clinical data, the safety and efficacy of the medicine have been satisfactorily established
- the quality of the medicine has been satisfactorily established, and
- they are satisfied with the sponsor's plan to submit comprehensive clinical data on the safety and efficacy of the medicine before the end of the provisional registration period (starting on the day that registration would commence)
These factors are outlined in subsection 25(1)(d) of the Act. Failure to meet these requirements during the registration process will provide us with a reason to consider rejection of the submission.
The provisional registration period for medicines that are approved will be two years (subsection 29(3) the Act). If required, you may apply for up to two extensions of up to two years each to extend the provisional registration period to a maximum of six years.
Appeals
Appeals of registration decisions are possible under section 60 of the Act. Please note that the decision whether or not to provisionally register a medicine is appealable by the applicant only and must be lodged within 90 days of the decision.
Further information on how to seek internal review by TGA or external review by the Administrative Appeals Tribunal is available on our website.
Phase 8: Post-decision
Provisionally registered medicines will be included in the part of the ARTG which is specific to provisionally registered medicines (section 6AAE of the Act).
The process for ARTG inclusion is the same as for the standard prescription medicine registration process.
These medicines will appear in the searchable ARTG on the TGA website and the Product Information and Consumer Medicine Information will be available on the TGA website in the same way as for any other registered medicine.
Provisionally registered medicines will also be subject to the Black Triangle Scheme.
Applications to extend the provisional registration for a further period will be considered in accordance with section 29 of the Act. Please see the guidance on provisional registration extension and transition to full registration for more information.
Clinical study plan for provisional registration
Example of clinical study plan overview for pivotal evidence at time of submitting an application for provisional registration
Example clinical study plan for provisional registration [Word, 37.71 KB]
Page history
Title changed from 'Provisional registration process' to 'Applying for provisional registration for a prescription medicine' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Minor updates and clarifications
Original publication
Title changed from 'Provisional registration process' to 'Applying for provisional registration for a prescription medicine' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Minor updates and clarifications
Original publication