Understanding the application requirements for an assessed listed medicine
Guidance on the regulatory requirements for assessed listed medicines.
Purpose
This guidance applies to 'assessed listed medicines' that are eligible for evaluation by our Complementary and Over-the-counter Medicines Branch (COMB).
There are 3 types of medicines that may be supplied over the counter in Australia based on the level of risk of the ingredients and therapeutic indications of the medicine:
- listed medicines
- assessed listed medicines
- certain registered medicines (such as registered complementary medicines).
There are 3 application categories for assessed listed medicines, with different evidence requirements based on the level of evaluation:
- L(A)1
- L(A)2
- L(A)3.
This document provides guidance for the evidence requirements for all 3 application categories, with a focus on the L(A)3 application category as these require a de novo evaluation of efficacy.
Legislation
Overview
The assessed listed medicines pathway offers sponsors a way to list products with low-risk ingredients in the Australian Register of Therapeutic Goods (ARTG) after we have pre-market assessed the efficacy evidence.
The pathway sits between the standard listed (lower risk) and registered (higher risk) pathways. It allows sponsors to list products with higher level indications than standard listed medicines without meeting the extensive requirements for registration, provided that the product has strong scientific evidence to support it.
The benefits of this pathway include the use of intermediate level indications, a label claim that we have assessed the efficacy of the product, and the potential for data protection.
Assessed listed medicines contain only permissible ingredients, meet the eligibility criteria for listed medicines, but are approved to use one or more intermediate level indications that are a higher level than indications in the Therapeutic Goods (Permissible Indications) Determination.
Assessed listed medicines can only be listed in the ARTG after the sponsor self-certifies the safety and quality of the medicine, and the TGA pre-market assesses the efficacy evidence supporting the indications and claims. Once approved by the TGA, they are given a unique AUST L(A) number and may carry a claim that the product’s efficacy has been assessed for the approved indications.
Risk-based classification
Like all listed medicines, assessed listed medicines may only contain low risk ingredients specified in the Therapeutic Goods (Permissible Ingredients) Determination and must be produced under Good Manufacturing Practice (GMP) principles. The safety and quality of the finished products are self-certified by the applicant and are not pre-market assessed by the TGA.
Assessed listed medicines may make indications that are higher risk or more definitive than indications in the Therapeutic Goods (Permissible Indications) Determination, but do not require registration. These ‘intermediate level’ indications are generally more definitive than indications for listed medicines, relate to more serious health conditions, and may lead to a delay in seeking medical treatment or adverse consequences if the product is used incorrectly or is not efficacious.
While assessed listed medicines are low risk based on their ingredients, manufacture, and route of administration, they are considered to be of higher risk than listed medicines based on the intermediate indications they carry. Thus, the TGA evaluates the scientific evidence supporting the indications before the product can be supplied.
Only products supported by high-quality scientific evidence are accepted through the assessed listed medicines pathway. Products that have indications supported solely by animal studies, tradition of use, or anecdotal data will not be accepted.
If the TGA determines that the efficacy of the proposed product is well supported by evidence and the product meets the other requirements for listed medicines, it can be listed in the ARTG.
Benefits of the assessed listed medicines pathway
The assessed listed medicines pathway bridges a significant gap for sponsors between the evidence requirements, costs, and timeframes in the listed and registered medicines pathways. It allows listed medicines, especially complementary medicines, to gain greater recognition of the efficacy of products that contain lower risk ingredients.
It provides opportunity to access higher level indications than are permitted for listed medicines, without requiring safety and quality data for the low-risk ingredients they contain. This pathway offers a market advantage via higher level indications, an ability to make a claim that the TGA has assessed the efficacy on labels and advertising, and the potential for data protection. This in turn supports innovation, competition, and an expansion of the evidence base for complementary medicines.
The pathway provides benefits to consumers and health professionals by increasing the transparency of the evidence base for therapeutic indications, improving confidence in products, enabling more informed healthcare decisions, and facilitating greater consumer access to a wider range of evidence-based medicines to self-manage their health.
The TGA has implemented a data protection scheme for assessed listed medicines to incentivise innovation and protect investment from competitors seeking generic forms of an L(A) medicine. For information about how to apply for data protection for the evidence provided to support an application, see the Data protection scheme for assessed listed medicines guidance.
Eligibility and regulatory requirements
To utilise the pathway, the proposed product must meet all the safety and quality requirements for listed medicines and meet specific requirements relating to efficacy and presentation.
Applicants must certify that the proposed product meets the requirements of subsection 26AB(2) of the Therapeutic Goods Act 1989 (the Act), and if applicable, subsection 26AB(3). Detailed information on the safety, quality and efficacy statutory requirements and conditions for listed medicines can be found in the Australian Regulatory Guidelines for Listed Medicines and Registered Complementary Medicines. The requirements for assessed listed medicines are similar to the requirements for listed medicines, except with respect to the premarket assessment of the efficacy evidence by the TGA.
Key elements of the assessed listed requirements are summarised below.
General requirements
Medicines listed via the assessed listed medicines pathway must:
- only contain permitted ingredients and meet the requirements for those ingredients specified in the Therapeutic Goods (Permissible Ingredients) Determination.
- not contain ingredients included in a schedule to the Poisons Standard
- not be required to be sterile
- not be a prohibited import for the purposes of the Customs Act 1901
- hold information showing the medicine’s specifications will be maintained according to the recommended storage conditions, until the expiry date on the medicine’s label
- comply with all applicable standards and legislative requirements in relation to quality and safety of medicines
- not be an export only medicine1 and
- not be a medicine that has previously had its registration or listing cancelled.
The sponsor must self-assess and certify under subsection 26AB(2) of the Act that the medicine meets the above requirements. These aspects of the medicine are not pre-market assessed by the TGA, however may be subsequently reviewed during standard post-market compliance processes.
If the medicine exceeds some of the above requirements (e.g. ingredients not in the Permissible Ingredients Determination), then it may be suitable for the registered pathway.
Evidence of GMP
Assessed listed medicines must be produced in accordance with the PIC/S Guide to GMP, which may be interpreted in accordance with listed and complementary medicine GMP guidance.
Applicants must provide valid evidence that the manufacturer(s) of the product have applied GMP for each step of manufacture included on the ARTG.
Evidence must be in the form of:
- Australian manufacturers: a copy of a GMP licence issued by the TGA
- overseas manufacturers: a GMP clearance issued by the TGA.
Applicants must ensure that the GMP clearance is valid prior to approval of the product. The evaluation timeframes vary for different application categories. If the GMP clearance is due to expire within the minimum timeframe or is likely to expire before the application is finalised, applicants should either apply to renew the GMP clearance or seek an extension to the GMP clearance.
For further information refer to Manufacturing medicines and GMP clearance for overseas manufacturers.
Indications and presentation
To be eligible for the assessed listed medicine pathway, the product:
- must carry one or more intermediate level indication(s)2 (an indication that exceeds the Therapeutic Goods (Permissible Indications) Determination but is not a high level indication)
- must carry the intermediate indication(s) on the product label3
- may carry optional additional low level indications, which meet the criteria for a low level indication (including, but not limited to those in the Therapeutic Goods (Permissible Indications) Determination). Evidence to support these low level indications must also be supplied and pre-market assessed by the TGA
- must not make any reference to a prohibited representation
- must not have an unacceptable presentation, as specified in the subsection 3(5) of the Act and 3(A) of the Regulations
- must have a label assessed and approved by the TGA, and which meets the requirements of the Therapeutic Goods Order No. 92 – Standard for labels of non-prescription medicines (TGO 92)
- must conform to every requirement relating to advertising specified in Part 5-1 of the Act and the Therapeutics Goods Advertising Code.
The indications and presentation of the product will be pre-market assessed by the TGA. The only indications that can be used on the label are those that are assessed by the TGA and included in the ARTG entry.
Sponsors must ensure that the information contained in the application is correct. An incorrect certification could result in the product being cancelled from the ARTG under the provisions of paragraph 30(2)(bab) of the Act. Sponsors are given an opportunity to respond before cancellation through a ‘proposal to cancel’ notice.
Indications
Risk categorisation
’Indications’ are statements that relate to the purpose or health benefit of the product. They are defined in the Act as ‘the specific therapeutic uses of the goods’. ‘Therapeutic use’, as it relates to medicines, is defined by the Act to include ‘use in or in connection with preventing, diagnosing, curing or alleviating a disease, ailment, defect or injury in persons, or influencing, inhibiting or modifying a physiological process in persons’. All indications on the product label or other advertising must be included in its ARTG entry.
The TGA has a risk hierarchy of indications for medicines that may be supplied without a prescription. This takes into account the health status and potential vulnerability of the target population; the seriousness of any conditions mentioned; and the probability that a consumer may delay seeking medical treatment based on an indication.
On the basis of these risk factors, indications are categorised into 3 levels of risk: low, intermediate, and high:
- Low level indications include indications for self-diagnosable, self-manageable, and self-limiting conditions where a delay in medical treatment would not be detrimental to the consumer. Low level indications may only refer to general health maintenance, health enhancement, prevention of dietary deficiency, or those that imply a benefit for a non-serious form of a disease or condition. Low level indications are used in listed medicines, and may also be used in assessed listed in addition to one or more intermediate level indications. Listed medicines may only use low level indications that are specified in the Therapeutic Goods (Permissible Indications) Determination, while assessed listed medicines may use these or specify their own.
- Intermediate level indications exceed the criteria for low level indications. They are generally more definitive, may relate to more serious health conditions, and may present higher risk to consumers than low level indications. These indications include references to prevention, cure or alleviation of non-serious forms of a disease, ailment, defect or injury. Although these conditions will generally be self-limiting, self-diagnosable and/or self-manageable, medicines with these indications may have the potential to lead to a delay in seeking medical treatment and/or adverse consequences for the consumer.
- Intermediate level indications can also refer to serious forms of a disease, ailment, defect or injury (i.e. restricted representations)[4]. These are indications that can be permitted on lower risk medicines in acknowledgement of a compelling health benefit from the use of products that refer to them. As such, in addition to demonstrating efficacy of the product, the public interest criteria need to be satisfied to allow a restricted representation to be used.
- High level indications are those that refer to the treatment, cure or prevention of a serious form of a disease (i.e. restricted representation) and there is a higher risk of delay of treatment. Products carrying high level indications are not suitable for listing, and must be registered in the ARTG following a complete assessment of their safety, quality and efficacy by the TGA. Note: Registered medicines may also contain ingredients or doses beyond those permitted by the ‘Permissible Ingredients’ list. For more information, please see Submitting an application for registered complementary medicines.
The risk hierarchy for indications aligns with the classification of a medicine as a listed, assessed listed, or a registered product. Note that registered medicines can contain any level of indication (however are usually high level indications), assessed listed medicines can only use intermediate and low level indications, while listed medicines can only use low level indications drawn from the Therapeutic Goods (Permissible Indications) Determination. This framework is summarised in Table 1 below.
| Low level | Intermediate level | High level |
|---|---|---|
Indications from the Permissible Indications Determination. A low level indication may refer to:
A low level indication must not:
| Indications that are between low (from the Permissible Indications Determination) and high level indications. Intermediate level indications may refer to:
Intermediate level indications may include those indications specified in a non-permitted indications list (if such a list is made). An intermediate level indication must not:
| Indications that are higher than low and intermediate level indications. High level indications refer to the prevention, alleviation or cure of a serious form of a disease, ailment, or injury. A high level indication must not:
|
See Indications for assessed listed medicines for examples of intermediate and low level indications.
How to structure an indication
Sponsors of assessed listed medicines construct and submit their own low level and intermediate level indications. These indications must be supported by appropriate evidence and must not be confusing or misleading regarding the therapeutic uses of the medicine. A valid indication needs to clearly describe the specific therapeutic use of the medicine and needs to describe a therapeutic action (e.g. relieves) and target (e.g. joint pain in moderate osteoarthritis).
An indication may also include details about the severity, population, and timeframes related to the therapeutic use, depending on the specificity of the evidence. The components of an indication are summarised in Table 2.
| Component | Explanation | Examples |
|---|---|---|
| Action | The action, effect, mechanism or benefit of the product. | Alleviates, prevents, cures, reduces, relieves, increases etc. |
| Action qualifier | Terms that ensure the action is suitable for the level of evidence the sponsor holds. They often specify effectiveness. | Helps, temporary relief of, etc. |
| Target | The physiological/ psychological factor or process; or disease, ailment, defect or injury. | Headache, muscle cramps and spasms, fever, pain, osteoarthritis, etc. |
| Target qualifier | Terms that ensure that the target is suitable for the evidence the sponsor holds. | Mild, moderate, specified symptoms, healthy/normal, excess, etc. |
| Indication qualifier | Terms that may provide information relating to the evidence held by the sponsor. This includes terms that specify a target population and/or times of use. | In the elderly, in sports athletes, in women, after strenuous exercise, etc. |
The typical structure of an indication is illustrated below.
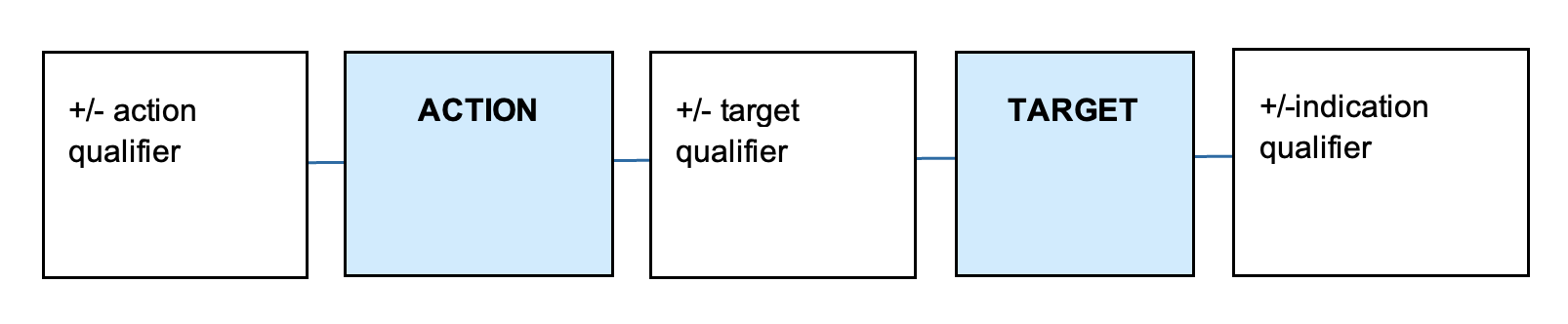
{kind=link}
A diagram consisting of 5 connected rectangles in a horizontal row. From left to right, the rectangles contain the following text:
- "+/- action qualifier"
- "ACTION" (in a light blue rectangle)
- "+/- target qualifier"
- "TARGET" (in a light blue rectangle)
- "+/-indication qualifier".
The rectangles are connected by horizontal lines, to suggest a sequence or relationship between these elements.
The diagram represents a structure or format for describing actions and targets with optional qualifiers.
Indications for assessed listed medicines
Intermediate (primary) indications
Assessed medicines must have at least one approved intermediate (primary) indication. Please note that:
- Intermediate indications can be linked to individual ingredients or the entire medicine, provided that the evidence for using the finished product supports the indications. Refer to the Evidence requirements and standards on how to support efficacy of finished product or individual ingredients.
- All intermediate indications must be supported by scientific evidence.
- Intermediate indications can imply clinical efficacy where they are supported by evidence.
- As intermediate indications are higher risk than those permitted for standard listed medicines, they must not be in the Therapeutic Goods (Permissible Indications) Determination.
Intermediate indications are classified into two categories:
(a) Indications for preventing, curing, or alleviating a non-serious form of a disease, ailment, defect, or injury.
These indications may refer to or imply the prevention, alleviation (relief or reduction of severity) or cure6 of a non-serious form of a disease, ailment, defect or injury. These indications may relate to:
- reduction in risk, frequency, duration, or severity;
- relief or reduction of symptoms; and/or
- complete resolution;
of a non-serious form of a disease, ailment, defect or injury.
Examples:
- ‘Prevents muscular cramps and spasms’
- ‘Alleviates mild dermatitis’
- ‘Prevents cold sores’.
(b) Indications in connection with alleviating a serious form of a disease, ailment, defect or injury.
These indications can refer to the use of the medicine in connection with alleviating a serious7 form of a disease, ailment defect or injury (e.g. a restricted representation).
These indications may relate to:
- relief or reduction of symptoms (or their severity), without implying resolution, cure, alleviation (reduction in severity) or prevention of the disease, ailment, defect or injury.
Examples:
- ‘Reduces symptoms of tinnitus’
- ‘Relieves rheumatoid arthritis symptoms such as pain and stiffness’
- ‘Relieves heartburn symptoms associated with gastroesophageal reflux disease’.
In contrast, the following indications may not be used as they imply resolution, cure, alleviation (reduction in severity) or prevention of the disease, ailment, defect or injury.
Examples:
- ‘Prevents type 2 diabetes’
- ‘Cures high blood pressure’
- 'Reduces the severity of gastroesophageal reflux disease’.
Vitamin D, calcium, or folate medicines
The Therapeutic Goods (Permissible Indications) Determination includes 3 indications referring to restricted representations for osteoporosis (for vitamin D and calcium) and neural tube defects (for folic acid). These indications have been permitted to be used in listed medicines based on the public health importance, safe history of use, and well-established evidence base of these substances. Listed medicines can use these indications without the need to transition to the assessed listed medicines pathway.
However, vitamin D, calcium, and folate products with indications that are not permitted in the Permissible Indications Determination, and all other indications that refer to a restricted representation, will require assessment and approval through a pre-market assessment pathway.
Low level (secondary) indications
Low level (secondary) indications drawn from (or consistent with) the Permissible Indications Determination may be included on assessed listed medicines in addition to the primary intermediate level indication(s). The secondary indications must meet the requirements for low level indications described in the Permitted indications for listed medicines guidance.
Sponsors must submit evidence to support all secondary indications and these are pre-market assessed by the TGA. The level of evidence required to support secondary indications is consistent with the requirements for standard listed medicines.
Secondary indications may:
- be associated with individual ingredients or the whole medicine provided that the evidence for the finished product and/or individual ingredients support the indications. If an indication is linked to a specific ingredient in the medicine’s formulation, then that ingredient should be linked to that indication on the medicine’s label;
- be general (non-specific) or specific in nature. General indications are those relating to health maintenance and supplementation or relief of symptoms not related to a named condition (e.g. ‘helps soothe dry skin’). Specific indications refer to named conditions or symptoms, health enhancement or specific therapeutic effects (e.g. ‘helps relieve indigestion’).
For assessed listed medicines, all indications must be supported by scientific evidence, and indications based solely on traditional use are not acceptable. However, traditional indications can be included as secondary indications if the therapeutic use they describe is also supported by scientific evidence. Additionally, for indications that were developed from a traditional paradigm, sponsors can provide truthful and verifiable claims, supported by evidence (see Claims).
Refer to the Supporting claims and indications for listed medicines (Section 4.2) for information on scientific and traditional indications that are suitable as secondary indications.
It is important to ensure that your indication aligns with the evidence you have to support it. The supporting studies generally need to refer to medicines with an equivalent formulation, preparation, dosage, and duration of use, and must have been carried out in a similar population group and context (refer to sections on alignment and presentation on this page for more information). Variation or extrapolation from the studies must be addressed with a robust scientific justification.
Application categories
Applications for assessed listed medicines are made under section 23B of the Act. There are 3 categories of application for new assessed listed medicines, L(A)1 – L(A)3 and categories for changes to an existing assessed listed medicine. The increasing levels correspond to the increasing complexity of applications, and consequently, increasing data requirements, evaluation timeframes and fees.
The data requirements for L(A)1-L(A)3 applications are outlined in the mandatory requirements for an assessed listed medicine application. You must meet these requirements to pass preliminary assessment and proceed to evaluation.
Application category: L(A)1
This category includes products that are identical to an existing assessed listed medicine other than permitted differences, such as the name, colour, printing ink, flavour and/or fragrance.
The following conditions must be met:
- the reference medicine[8] must have been fully evaluated for efficacy by the TGA
- the reference medicine must comply with all current requirements and standards, relevant Therapeutic Goods Orders (e.g. TGO 92, TGO 95, TGO 100, TGO 101) and default pharmacopoeial standards
- the label, indications, and formulation must reflect the fully evaluated reference medicine
- full access by the TGA to the reference medicine dossier must be provided. The sponsor of the reference medicine must authorise the TGA to access the information on the reference medicine files and ARTG record for the purpose of the application.
Permitted differences
Table 3 summarises the list of permitted differences for L(A)1 applications.
| Difference | Description |
|---|---|
| Sponsor details | The sponsor of the flavour/fragrance/colour variant can differ from the sponsor of the reference medicine, provided the sponsor of the reference medicine authorises the TGA to access the information on the reference medicine files and ARTG record for the purposes of the application. |
| Medicine name | The proposed medicine name must be different to the reference medicine. The proposed medicine name cannot include a claim or indication that is not approved for inclusion in the ARTG entry or name of the reference medicine. Including a subset of the approved indications in the medicine name is not permitted. |
| Manufacturing sites | The manufacturing sites for the proposed medicine can differ from the reference medicine where the proposed manufacturer has been validated and shown to be equivalent or better. Ensure that you have valid evidence of GMP for the manufacturers. |
| Flavour/fragrance/colour variants | Only the flavour, fragrance and/or colour agents can differ in the proposed medicine, and the combined total difference cannot be more than 2% w/w or w/v of the total formulation. The proposed raw material specifications for new flavour/fragrance/colour must comply with applicable standards, and all components are included in the Therapeutic Goods (Permissible Ingredients) Determination. If the new flavour/fragrance/colour includes excipients that must be declared on the label, ensure that the labels have been updated accordingly. If the medicine label includes any claims that it does not contain a particular excipient (e.g. gluten free, sugar free, lactose free) ensure the claims are true in regard to the components of the new flavour/fragrance/colour. |
| Pack size | The proposed pack size(s) can differ from the reference medicine only for solid dosage forms where there is no change in container material. |
| Medicine labels | The labels for the proposed medicine must be identical to the reference medicine, other than the medicine name, design and layout, pack size details, sponsor or supplier details and logos. The proposed label graphics can differ from those approved for the reference medicine provided that:
|
| Finished product specifications/visual identification | The finished product specifications must be identical to those approved for the parent medicine other than the flavour/fragrance/colour (including printing inks) aspects. The visual identification can differ from the reference medicine only when it is either a direct consequence of the new flavour/fragrance/colour agent(s); or a difference in debossing/embossing/printing to remove or add identifying marks. |
Application category: L(A)2
This category is for generic medicines or medicines where a Comparable Overseas Body (COB) report has been provided to support efficacy.
Generic medicines
In comparison to the fully TGA evaluated assessed listed reference medicine, the proposed generic medicine9 must:
- have the same active ingredient(s) with same quantity and similar quality
- have the same pharmaceutical form
- be bioequivalent to the reference medicine (meet the Biopharmaceutic and pharmacokinetic requirements)
- have the same safety and efficacy properties10.
The generic medicine should provide a justification of the use of the particular combination of ingredients including potential interactions.
Medicine evaluated by a Comparable Overseas Body (COB)
The TGA’s COB report-based process allows technical evaluation reports from identified bodies to be used by TGA to assess applications against the Australian requirements. For the current list of COBs and guidance for using COB evaluation reports, refer to Comparable overseas bodies (COB).
Application category: L(A)3
This category includes all products that are not covered by L(A)1 or L(A)2 and is either:
- a new medicine requiring a de novo evaluation of the efficacy of the product
- a change to an existing approved assessed listed medicine, where the medicine has one of the following:
- different active ingredient(s)
- different strength (i.e. quantity of active ingredient(s))
- different indication(s) (other than removing an indication)
- different dosage form
- different excipients.
L(A)3 applications must provide complete Module 1, Module 2 and Module 5, as applicable.
To demonstrate efficacy, L(A)3 applications must:
- provide efficacy evidence in line with Table 5 (using Method 1, 2A or 2B)
- meet the evidence requirements specified under Evidence requirements and standards and Alignment of indications and evidence.
Changes to an existing assessed listed medicine
Once a medicine has been listed in the ARTG, the sponsor may request approval to make a change to the medicine or correct the ARTG entry whilst retaining the existing AUST L(A) number. For more information about the change categories and data requirements see Changing a listed or assessed listed medicine: application levels and change tables.
Applications to change existing assessed listed medicines must be made via the assessed listed medicine form in TGA Business Services (TBS) system.
Evidence requirements and standards
This guidance provides information on the general types and standards of evidence required to support an application for an assessed listed medicine. Refer to application categories for specific data requirements.
Methods of establishing efficacy
Overview of methods of establishing efficacy for different application categories
Each application category has different methods for establishing efficacy of the product. These methods are generally summarised in Table 4 below. The methods of establishing efficacy for L(A)3 applications are described in more detail in Table 5.
| Application type | Product type | Method of establishing efficacy |
|---|---|---|
| L(A)1 | Identical to an existing assessed listed medicine, other than a permitted difference as specified in Table 3. |
|
| L(A)2 | Generic medicine of a fully evaluated assessed listed medicine. |
|
| A medicine that has been fully evaluated by a comparable overseas body (COB). |
| |
| L(A)3 | Any type of product. |
|
| Isolated chemical substances (i.e. single chemicals, well-defined chemical complexes, prodrugs, amino acids, vitamins and minerals). |
| |
| Products that meet the requirements for a compliant biowaiver and medicines that do not require biopharmaceutic studies or clinical efficacy studies (e.g. some aqueous oral solutions or some products containing substances that are not systemically or locally absorbed). |
|
Methods of establishing efficacy for L(A)3 applications
All indications for assessed listed medicines must be supported by scientific evidence of efficacy of the product. It is important to note that efficacy is not the same as effectiveness:
- Effectiveness is the extent of perceived or reported beneficial effect under ’real world’ settings, and may be different than efficacy as a consequence of factors that are controlled or limited in clinical settings but not in real world use (e.g. different population groups, diets).
- Efficacy relates to the extent to which an intervention produces a beneficial effect under ideally controlled conditions, such as in a randomised controlled trial. The goal of efficacy studies is to determine the causal relationship between a treatment and the observed effect.
While medicines eligible for the assessed listed medicines pathway may operate through different therapeutic modalities to conventional medicines, the method of assessing outcomes should be scientifically valid. Similar principles and standards of efficacy evidence apply to these products.
For L(A)3 applications, there are 3 methods via which applicants may provide evidence of the efficacy of the proposed product (see Table 5). These 3 methods are designed to ensure that there is a sufficient standard of evidence to support consumer confidence in the indications, while being sufficiently minimal to enable access to the pathway and support innovation in the sector. In brief:
- Method 1 utilises the common standard approach of clinical trials on the finished product, and is suitable for all product types. More complex formulations such as herbal medicines and probiotics would normally use this approach.
- Method 2A uses ingredient efficacy data underpinned by bioavailability/bioequivalence data (or a suitable justification) to support the plausible efficacy of the finished product. It is usually suited to systemically acting isolated chemical substances (i.e. single chemicals, well-defined chemical complexes, prodrugs, amino acids, vitamins and minerals).
- Method 2B uses ingredient efficacy data underpinned by product dissolution/release data or in vivo pharmacokinetic studies to support plausible efficacy of the finished product. It can only be used for products that meet the requirements for a compliant biowaiver[11] and certain medicines that do not require biopharmaceutic studies or clinical efficacy studies (e.g. some aqueous oral solutions or some products containing substances that are not systemically or locally absorbed)[12] See Biopharmaceutic and pharmacokinetic studies for further information.
Table 5 specifies the minimum requirements and may vary depending on the product.[13]
| Data type | Method 1 | Method 2A | Method 2B |
|---|---|---|---|
| Suitable product types | All types | Systemically acting isolated chemical substances. | Supported by a biowaiver, or not requiring biopharmaceutic or clinical efficacy studies. |
| Literature search report demonstrating body of scientific information[14] | Full literature search report on the finished product[15], or all active ingredients and formulation. | Full literature search report on all active ingredients and formulation. | Full literature search report on all active ingredients and formulation. |
| Published studies or clinical study reports (see Table 7) | Efficacy evidence on the finished product[16]. | Efficacy evidence for each active ingredient. | Efficacy evidence for each active ingredient. |
| Biopharmaceutic and pharmacokinetic evidence | Not normally required | Evidence for efficacy of the product formulation, established through:
| In vitro dissolution/ release tests or pharmacokinetic studies demonstrating in vivo drug release and availability of the active ingredients at the site of action. Scientific justification of the approach and validation of the approach where appropriate. |
| Formulation | All methods must provide a justification (rationale) for the particular combination of ingredients in the finished product, including potential interactions between the ingredients. | ||
Note that Methods 2A and 2B are generally not suitable for herbs, herbal extracts, substances of biological origin, or complex mixtures of chemicals. This is because the variable chemical composition and, in many cases, lack of known active component, makes it difficult to accurately demonstrate appropriate biopharmaceutical properties of the medicine. A sub-set of chemical markers is not a suitable proxy for establishing the bioavailability or bioequivalence of all active constituents of a complex substance.
Applicants may submit a detailed scientific justification if the data package demonstrates that bioavailability/bioequivalence data is not required (see Justifications).
Evidence based on literature of the active ingredients (not necessarily a clinical trial conducted on the finished product that is the subject of the L(A) application) can be used for Methods 2A and 2B provided the evidence is robust and supported with biopharmaceutic and pharmacokinetic evidence or a suitable justification that it is bioavailable.
Products that do not meet the evidence requirements for Methods 2A and 2B may either be assessed via Method 1. Alternatively, such products may be listed via the listed medicines pathway and use indications in the Permissible Ingredients Determination.
Types of applications
Some of the above evidence requirements for L(A)3 applications can be met through:
- Conventional applications - primarily contain full study reports of company sponsored studies that support the efficacy of the product. These studies can be supported with bibliographical references.
- Literature-based submissions - rely on bibliographic data or overseas reports to support the efficacy of the product (See Using literature-based submissions for listed, assessed listed and registered complementary medicines). The literature must be relevant to the application e.g. the information should relate closely to the formulation, dosage regimen and indications of the proposed product. Unlike for prescription medicines, you do not need to gain approval of literature search strategies prior to submitting your application, and there is no formal pre-submission phase.
- Mixed applications - consist of a combination of full study reports of limited clinical studies carried out by the applicant supplemented with bibliographical references to support the efficacy of the product.
Regardless of the approach used, a certified translation should be provided for relevant evidence reported in a language other than English. All evidence will be subject to minimum requirements for relevance, quality and consistency.
If applicants utilise a literature-based submission or mixed submission, all scientific publications should be peer-reviewed and be published in a reputable journal.
Table 7 outlines the evidence requirements for primary and secondary indications.
Literature search report
A literature search report is a description of a logical, transparent and reproducible approach to identifying and retrieving all authoritative published material which contains evidence (both positive and negative) related to the proposed product and/or its components. It is intended to provide a comprehensive and unbiased review of the available literature in relation to the application, and is a key requirement of all evidence-based medicine.
All applicants must include a report of the methodology used for the systematic literature search with the application in Module 1.5.1. The report should include, as a minimum, a well-conducted systematic search of Medline or Embase with descriptions of any additional non-systematic or manual searching. The report must outline:
- the search strategy, rationale, platform and date
- references retrieved and period covered
- selection or filter criteria applied to identify relevant reports
- list of reports which have been excluded
- appraisal of the evidence identified
- pivotal studies and the rationale for their selection
- details of how any additional references were retrieved - for example, from in-house databases, lists of references, or hand searching.
For a search strategy to be considered robust it should be reproducible. Applicants should not substitute 'in-house' databases for Medline and/or Embase searches or use internet search engines as a primary search platform.
However, applicants may include other appropriate public databases in addition to Medline/Embase, and must include all relevant studies regardless of whether the findings are adverse to the proposed product or not. Relevant reports include all studies that reference (amongst others) the product ingredients, formulation, dose, health benefit, and context of use.
No single literature search strategy will fit all cases and requirements will vary according to the specific nature of the application.
In planning and conducting systematic literature searches, you may find it useful for an information retrieval expert to be involved in the process.
For further guidance on conducting literature searches, please refer to the listed medicine evidence guidelines (Section 2.2). For more information on documenting literature searches, please refer to guidance on systematic literature searches.
Standards of evidence
The evidence provided to support an application for an assessed listed medicine should cover aspects of the pharmacology, clinical safety (e.g. relating to the dose, use in vulnerable populations, specific formulation and dose form), and efficacy of a medicine, and serve to establish the balance of benefits and risks of the medicine in relation to its intended use. It should also provide the scientific evidence to support the claims and directions for use made on product labels and other product literature.
In assessing the evidence, the TGA must be assured that outcomes observed are due to the therapeutic action of the product and are not simply due to chance or sources of experimental bias introduced by the design, execution, or reporting of the study. The outcomes should show a clinical benefit, and plausibly applicable if indicated for a wider population.
Overall, the standard and weight of evidence submitted for an assessed listed medicine should support a plausible cause-effect relationship between the treatment or intervention and the presumed therapeutic outcome. To ensure that such inferences can be made, the standard of evidence submitted by applicants is reviewed based on:
- the type/ design, and quantity of evidence)
- internal validity (i.e. methodological quality)
- statistical validity
- external validity (generalisability)
- extent of evidence consistency
- relevance of the evidence to the product and indications.
Further detail about the quality of efficacy evidence can be found in Appendix A.
Evidence hierarchy and requirements
The TGA takes a ‘weight of evidence’ approach - the less robust the studies, the greater the quantity of consistent evidence required. To assist sponsors to ensure that they have at least the required minimum of appropriate level of evidence to support their application, the TGA has developed an evidence hierarchy and minimum evidence framework. These are provided in Tables 6 and 7. The definitions for intermediate and low level indications have been provided previously (see Risk categorisation).
Note that the requirements in Table 7 are generally minimum evidence requirements and the options presented may not be suitable for every situation. This represents the lower threshold below which the efficacy of the medicine cannot be reasonably assessed. Supplying the data in Table 7 is not sufficient for an application to be approved. The information must be of high quality and address the other requirements that address the efficacy of the product as set out in this document (see Evidence requirements for efficacy evaluation and Alignment of indications and evidence). Information on specific areas of concern may need to be addressed through more or better quality studies.
It is not mandatory for applicants to rely on one pivotal study. However, applicants who choose to rely on one pivotal study on the finished product as the primary supporting evidence should meet the TGA-adopted EU guidance document Points to Consider on Application with 1. Meta-analyses; 2. One Pivotal Study. The guideline sets the minimum requirement of generally one controlled study with statistically compelling and clinically relevant results with high data quality.
Additional studies, where available, serve to strengthen the evidence of efficacy, particularly if any of the primary evidence is limited in some way, and may improve the likelihood of an application being approved.
Regardless of what studies are used, all sources of evidence must meet acceptable standards of evidence and include an adequate description of the study design, population, treatment(s) and protocols employed for evaluation purposes. As such, abstracts, web searches or incomplete references will not be accepted as suitable evidence.
| Category A | Category B | Category C | Category D |
|---|---|---|---|
| Double blind randomised controlled trials (including cross-over trials) | Observational studies e.g. cohort and case control studies | Non-systematic, generalised reviews - including databases | Traditional reference text |
| Systematic reviews or meta-analysis of randomised controlled trials | Intervention studies (non-control) | Publicised international regulatory authority articles | Herbal monograph |
| Evidence based reference text - scientific | Herbal pharmacopoeia | ||
| Scientific monographs | Materia medica | ||
| Publicised international regulatory authority articles – Traditional only |
| Indication | Primary (intermediate) | Secondary | |
|---|---|---|---|
| Indication type | Scientific | Scientific | Traditional[17] |
| Required evidence | Minimum of one from Category A OR Minimum of two from Category B, AND one from Category C | Non-specific indications: Minimum of two from Category B or Category C | Non-specific indications: Minimum of two from Category D to support the tradition of use |
Specific indications: Minimum of one from Category A OR Minimum of one from Category B, AND two from Category C | Specific indications: Minimum of two from Category D to support the tradition of use Plus Additional evidence from Category C or Category D to support the specificity of the traditional indication | ||
Double blinded randomised controlled trials, and systematic reviews and meta-analysis of multiple randomised clinical trials are the gold standard in epidemiological and clinical research, as they are most likely to achieve low bias and high precision when studying treatment effects. However, they are not always available or feasible. Acknowledging this, the TGA allows other study types and a range of other sources of evidence to be submitted as potential support for the claimed efficacy of a product. The limitations of these other sources need to be considered.
- Well-conducted clinical trials can be used as evidence to support both intermediate level and low level indications. Clinical trials submitted as evidence to support an indication should be published in a high quality, peer-reviewed journal. All published and original unpublished clinical trials should meet all the applicable international scientific guidelines adopted in Australia.
- In general, robust systematic reviews and meta-analysis can be used as evidence to support both intermediate level and low-level indications.
- Cohort and case control studies are limited in their ability to provide unbiased and unambiguous data regarding the true efficacy of an intervention, and therefore may not provide acceptable evidence for some indications.
- Non-controlled intervention studies are often limited by an inability to distinguish between the effect of the treatment, a placebo effect, and the effect of natural history. Additionally, they may fail to identify positive effects in situations where a negative outcome would have resulted in the absence of the intervention. They are, therefore, not appropriate as a sole source of evidence for the efficacy of an assessed listed medicine, but can be used as supporting evidence in combination with other sources of evidence.
- Although informative, non-systematic reviews are limited by selection and author bias. They may also fail to provide clear conclusions, particularly if the studies included have conflicting results. Due to this, non-systematic reviews cannot be used as a sole source of evidence for efficacy, but can provide support for other studies.
- Several internationally recognised monographs and reference texts are available and may be used to support secondary (low level) indications. Only sources that include scientific/ clinical information are appropriate to support secondary scientific indications.
- While all indications must be supported by scientific evidence, if your application has indications that refer to use in a traditional context (these can only be low level indications), then it may use traditional evidence to support the context of traditional use. Some examples of sources of evidence for traditional use can be found in Appendix 1 of the Listed medicine evidence guidelines.
The types of evidence for supporting scientific indications can be found in Oxford Centre for Evidence-Based Medicine: Levels of evidence.
Only human studies are considered appropriate to support indications for assessed listed medicines. The scientific uncertainties involved in extrapolating non-human data from animal and in vitro studies limit their usefulness. However, non-human and in vitro studies may be used to support any discussion on biological plausibility, and in vivo and in vitro studies may be used when providing biopharmaceutic and pharmacokinetic data.
Non-reference textbooks, web searches, and publication abstracts are not appropriate sources of evidence to support an application for an assessed listed medicine.
The Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach may be helpful in assessing the overall certainty in the evidence. The GRADE assessment provides a structured way to consider key factors that may increase or decrease our confidence in the synthesised findings of a body of evidence. Further guidance can be found in the GRADE working group.
To meet the above minimum evidence requirements, the evidence should contain independent sources of information e.g. two publications referencing the same clinical trial or information are not considered to be two independent sources of information.
Biopharmaceutic and pharmacokinetic studies
Biopharmaceutic and pharmacokinetic studies are a critical part of establishing the efficacy of medicines. These types of studies demonstrate that medicines release active ingredients appropriately; and that the active ingredients are absorbed, distributed, and metabolised in a manner that allows the active ingredient to reach the intended site of action. They also serve to ensure that undesirable effects such as dose-dumping, dose retention or in vivo interactions do not either reduce the efficacy of the product or pose a risk to the consumer.
For L(A)3 applications (using Method 2A or 2B) and L(A)2 generic applications, different biopharmaceutic and pharmacokinetic studies are required to ensure that the products are likely to be efficacious. The data requirements are summarised below depending on the type of product. For more information, see Providing data for prescription medicine applications[18]
Pharmacokinetic data is not explicitly required when a clinical study is used as the main evidence for an intermediate indication (Method 1), however such studies will generally, where necessary, address relevant aspects of the medicine’s pharmacokinetic properties.
New products (systemically acting)
For L(A)3 applications to establish the efficacy of a new systemically acting product, applicants may need to provide relevant data or information on bioavailability. This is particularly important in the case where the evidence is derived from efficacy studies on the individual ingredients (Method 2A), however as described in Table 5, is not usually necessary for Method 1.
Bioavailability is the proportion of the administered dose of an active ingredient that reaches its intended biological destination, such as the systemic circulation. It may differ between individuals and depends on a large number of factors that cannot usually be reliably inferred from the formulation. For systemically acting oral products these include the:
- rate and extent of the disintegration of the product, and the rate and extent of dissolution of the active ingredient(s)
- rate and extent of passage of the active ingredient through the gut membranes – a process determined by factors such as the physiochemical characteristics of the ingredient, including its lipid solubility, diffusivity and propensity towards interactions with active transporters in the gut wall, as well as the excipients used in the formulation, the drug coatings, and the gut lumen pH
- rate of gastric emptying/ intestinal transit
- extent of first-pass metabolism in the liver – if first-pass metabolism occurs, some proportion of the substance will be removed before the remainder reaches systemic circulation.
For these reasons, some applications may need to address several factors that impact on the efficacy of the medicine. These requirements also differ depending on whether the product is intended to be an immediate release product or a delayed/modified release product.
Immediate release oral dosage forms
The following studies (or a robust scientific justification for not including such studies) are required:
- study to establish that the proposed formulation is optimal (e.g. a comparative bioavailability study versus an oral solution of the drug)
- bioequivalence studies between the proposed formulation and pivotal clinical trial formulations
- bioequivalence studies amongst the various strengths proposed in the application (if applicable)
- a food effect study.
Modified release oral dosage forms
Modified release products (including delayed, sustained, and combination release products) must be determined to meet the modified release claims; should provide consistent pharmacokinetic performance between dosage units; and should produce plasma concentrations that lie within the therapeutic range.
The following studies (or a robust scientific justification for not including such studies) must be submitted, in addition to the studies required for immediate release oral dosage forms:
- steady state versus an appropriate immediate release reference product
- in vitro and in vivo correlation studies
- in vitro studies confirming the absence of dose-dumping effects in the presence of alcohol.
Generic products
For L(A)2 and in some cases for L(A)3 applications, the proposed assessed listed medicine may be similar or identical to an existing product for which bioavailability data exists. This includes products currently approved as an assessed listed medicine in Australia, evaluated by comparable regulatory authorities, or extensively studied in clinical trials, and excludes ‘grandfathered’ products.
In such cases, the efficacy of the proposed product can be inferred if it can be demonstrated that it has the same / equivalent pharmacokinetic properties or bioavailability to the reference product.
A generic assessed listed product is a medicine that, in comparison to another fully TGA evaluated assessed listed medicine (the ‘reference product’) included in the ARTG:
- has the same quantitative composition of therapeutically active substances, being substances of similar quality to those used in the comparison medicine; and
- has the same pharmaceutical form; and
- is bioequivalent; and
- has the same safety and efficacy properties.
All applications for generic systemically acting products must establish bioequivalence between the reference and the proposed products. Bioequivalence refers to the comparability of medicines that are pharmaceutically equivalent and which have no significant difference in the rate and extent to which the active ingredient becomes available at the site of drug action when administered at the same molar dose under similar conditions.
Bioequivalent drugs are similar to such a degree that their effects, with respect to both efficacy and safety can be expected to be essentially the same (see Providing biopharmaceutic data for prescription medicine applications (TGA Guidance).
Bioequivalence studies should be performed on the innovator product (i.e. a product that has had a full efficacy data package evaluated by the TGA), not a generic of the innovator. This is important to reduce the likelihood of pharmacokinetic drift, whereby generics that refer to other generics no longer resemble the originally evaluated product due to variability in confidence intervals in each bioequivalence study.
The most reliable means to demonstrate that one formulation will be as effective as another is to conduct a randomised, single-dose crossover bioequivalence study in healthy volunteers. In these studies, subjects receive the different formulations on two separate occasions separated by a wash-out period. A minimum of 12 volunteers, sufficiently long wash-out period (5-7 times the half-life of the drug) to prevent carryover effects, and adequate plasma sampling frequencies should be used.
Studies on subjects in the fasted state are usually preferred, as this is the most sensitive condition for detecting differences between formulations. In general, the 90 % confidence interval of the ratio of the geometric means of the area under the plasma concentration vs. time curve (AUC) and maximum plasma concentration (Cmax) are required to be between 0.8 and 1.25 for bioequivalence to have been demonstrated.
There are two ways to demonstrate bioequivalence:
- where the reference product has previously not been evaluated for bioavailability, bioavailability studies of both the reference and the proposed product are required
- where the reference product has been evaluated for bioavailability by the TGA, similar dissolution profiles between the reference and the proposed products across the physiological pH are taken into consideration to establish bioequivalence.
If the formulations differ significantly and a different release rate has been designed into the formulation, then a non-inferiority study against the reference product may be appropriate.
More information:
- Providing biopharmaceutic data for prescription medicine applications
- The European Medicines Agency (EMA) Investigation of bioequivalence - Scientific guideline
- The EMA Pharmacokinetic and clinical evaluation of modified-release dosage forms - Scientific guideline.
Generic products that have the same excipients as the reference product
If a product is a generic of an existing product for which bioavailability data exists, and has the same excipient formulation, applicants can demonstrate bioavailability by providing:
- evidence of identical formulations including excipients
- demonstration of similar dissolution profiles between reference and proposed products across physiological pH (where applicable).
Applicants may rely on bioavailability data for an existing product, or data that is obtained through literature, with the appropriate justification. The reference product cannot be a ‘grandfathered’ medicine.
Generic products meeting the requirements for a BCS-based biowaiver
In certain circumstances, despite the product being of a type that would normally require biopharmaceutic studies, it is possible to provide a robust scientific rationale for why bioavailability and/or bioequivalence data might be considered unnecessary for listing of the proposed product. This is generally referred to as a ‘biowaiver’.
Biowaivers allow dissolution tests to be used as the surrogate basis for the decision as to whether two products may be considered to be equivalent. In this context, the dissolution and absorption of the medicine is regarded as the critical aspect in determining the equivalence of two products. Consequently, biowaivers are only appropriate for certain classes of products.
The Biopharmaceutics Classification Scheme (BCS) is generally used to determine whether or not a biowaiver may be appropriate. The BCS classifies active substances into four classes based on solubility and permeability, as follows:
| High solubility | Low solubility | |
|---|---|---|
| High permeability | BCS class I | BCS class II |
| Low permeability | BCS class III | BCS class IV |
Products containing active(s) that are highly soluble, highly permeable (i.e. BCS class I substances) and rapidly dissolving may be considered for a biowaiver. Highly soluble substances are soluble at the highest dose strength in <250 mL water over a pH range of 1 - 7.5. Highly permeable substances are those for which the extent of absorption is > 90 % of an administered dose based on mass balance or relative to an intravenous reference dose.
Rapidly dissolving products are defined as those where no less than 85 % of the product dissolves within 30 mins in standard conditions. Excipients that might affect bioavailability should be qualitatively and quantitatively the same in the two products. In general, the use of the same excipients in similar amounts is preferred.
Biowaivers are not usually supported for BCS II – IV substances, or products with more complex formulations such as prolonged release tablets. In some instances, a BCS-based biowaiver may be considered for BCS III products (high solubility, low permeability) where the substance has high solubility and limited absorption; very rapid in vitro dissolution; excipients that might affect bioavailability are qualitatively and quantitatively the same; and other excipients are qualitatively the same and quantitatively very similar.
For additional information about the requirements for demonstrating that a substance can be considered to be BCS class I or III for the purposes of a biowaiver, refer to Appendix III of the TGA-adopted EMA Guideline on the investigation of bioequivalence. The FDA guidance on the BCS may also be helpful.
In all cases, the reference product to which the proposed assessed listed medicine is being compared must have established bioavailability data (e.g. assessed listed or registered medicines, apart from ‘grandfathered’ products, or products extensively characterised by overseas regulatory authorities).
The applicant’s justification should address all the aspects of the products outlined in Providing biopharmaceutic data for prescription medicine applications. Additionally, dissolution profiles across physiological pH showing appropriate release of the active(s) must be supplied (this is a standard quality requirement for all medicines).
Products not requiring biopharmaceutic studies
For some L(A)2 and L(A)3 applications, there is a limited number of products that do not require biopharmaceutic studies, even in the absence of a reference product.
These include:
- Aqueous oral solutions that contain the same active substances in the same concentration as a current evaluated product, and that do not contain any excipients that might affect the in vivo solubility, in vivo stability, gastric passage or absorption of the active ingredient(s). Refer to the Guideline on the investigation of bioequivalence for more information.
- Oral medicines that are not systemically or locally absorbed (e.g. probiotics, non-digestible polymers, oral suspensions etc.).
- Locally applied products where the active(s) are not systemically or locally absorbed.
- Products with only minor formulation changes - i.e. up to 2% of the total formulation can change compared to the reference formulation, if the change is only to a flavour, fragrance and/or colour (including printing inks). However, you may need to provide dissolution profiles across physiological pH showing appropriate release of the active(s).
- For variations to formulation; medicines with an acceptable correlation between the rate and extent of in vivo absorption and the in vitro dissolution rate, and where the in vitro dissolution rate of the reformulated medicine is equivalent (under the same test conditions used to establish the correlation) to the approved AUST L(A) medicine.
For products that are not systemically or locally absorbed, you may need to provide evidence of non-absorption, and efficacy may need to be demonstrated via clinical studies or other data. Please refer to the relevant guidelines e.g. Equivalence studies for the demonstration of therapeutic equivalence for locally applied, locally acting products in the gastrointestinal tract - Scientific guideline.
Justifications
In some cases, it might be unfeasible or scientifically unrealistic to supply some required evidence or to meet some of the guidelines mentioned in Sections 4 - 5 above. In such instances, applicants are able to submit a scientific justification (see more information on justifications in the Mandatory requirements for an assessed listed medicine application to pass preliminary assessment).
Alignment of indications and evidence
Regardless of how scientifically sound a study is, its suitability as evidence will depend on how closely the proposed indications for the product match the results and conclusions of the study.
The indications used on a product and the supporting evidence must:
- refer to the same medicine or active ingredient(s)
- refer to the same therapeutic action and context (e.g. target population).
Indications must also remain valid for the entire life cycle of the medicine.
Formulation and use
Applicants must ensure that there is concordance between the parameters in the efficacy evidence and the product. The product used in the evidence and the intended product should generally have the same:
- ingredient(s)
- dosage
- dosage form
- route of administration
- frequency and duration of use
- target population.
If there are differences, further evidence and justifications are required to address the data gaps (refer to Biopharmaceutic studies).
When evidence relates to a complex naturally occurring substance (herb, herbal extract, probiotic, animal-derived product), the species, sub-species, strain, parts, quantity of a known active component of an ingredient, and preparation should be identical to that described in the clinical trial. Any deviation will require a robust justification.
Given that the chemical profile of any complex substance can vary (e.g. herbs sourced at different times of the year, or from different climactic / geographical situations, may provide differing amounts of extractable herbal components), it is unlikely that deviations from the formulation described in a clinical trial will be accepted.
However. where there is known unavoidable natural variability, efforts should be made to reduce variability, for example, through standardisation of extracts or agricultural means.
Substituting equivalent herbal extract ingredients in listed medicines may be a helpful resource to guide sponsors on what to consider when formulating products and determine if a herbal extract is ‘equivalent’ to the proposed product.
Duration of studies
Studies must be of an appropriate duration for the indication or claim. The required duration will depend on the nature of the health benefit but must be sufficient for that benefit to be clearly demonstrated. For example, products that claim a six-week reduction of pain should run a study of at least 6 weeks.
However, products with long-term health benefits should be supported by studies of sufficient duration for a sustained response to be apparent. In some cases, long term benefits can be extrapolated using other evidence such as epidemiological studies, other clinical trials on similar products, and a discussion on physiological mechanisms to demonstrate biological plausibility.
Regarding indications for risk reduction and modulation of biomarkers, research should consider the role of homeostatic mechanisms and whether they may affect changes in the longer term.
Outcomes
Indications should reflect the primary outcome of a study with an adequate sample size, however indications based on secondary outcomes may be acceptable in some cases where these outcomes are statistically and clinically meaningful.
Regardless of the level of the evidence, the indications must not:
- exaggerate the extent, nature, or prominence of the effects achieved in a study; or
- suggest greater scientific certainty than the study is capable of providing; or
- imply efficacy in all instances.
Evidence describing the biological effect, rather than the clinical effect, is not generally a suitable basis for an intermediate indication on its own - although it may contribute to establishing the biological plausibility for the indication.
This is particularly pertinent in the case of indications that refer to the favourable modulation of biomarkers (e.g. blood glucose levels, cholesterol levels etc.). A small change in a given biological surrogate may be associated with negligible clinical outcomes or increases in risk. However in some cases, changes in biological surrogates may be used with a justification that they are reasonably able to predict clinical benefit in relation to supporting the indications.
Context
Efficacy studies are usually conducted under tightly controlled conditions in order to control for confounding variables. Studies conducted in this way are ideal for estimating potential efficacy but may not reflect effectiveness within its target population e.g. due to different population groups, diets, etc.
Applicants should therefore carefully assess the aspects of the study that were controlled and establish whether the absence of these controls in ‘real use’ are likely to impact on the benefits experienced by consumers.
For example, a study showing a weight loss benefit of a substance may control all participants’ caloric intake. Consequently, it is unlikely that the same benefits would be experienced in situations where caloric intake was not controlled.
In such cases where behaviour or other factors can act as major confounding variables, contextual qualifiers should be included in an indication or as an advisory statement to reflect the evidence base.
Example
‘Assist with weight loss when used with a calorie-controlled diet and exercise’.
Target population and generalisation/extrapolation
The target population for the medicine should be consistent with the population described in the evidence source unless extrapolation can be justified.
For the results of a study to be generalisable to the Australian context, the study used to support the indications for an assessed listed medicine should be conducted using a sample population that:
- consists of both female and male participants
- consists of individuals aged 18-65 years
- consists of healthy, or only mildly unwell, individuals
- is demographically similar to the Australian population.
Additionally, indications should not extrapolate or generalise the outcomes of a study to populations that differ significantly from that used in the study. Specifically:
- It is not appropriate to use studies carried out on populations with significant health concerns to support an indication for assessed listed medicines; unless the indication relates directly to a population with a serious condition (i.e. a restricted representation). If the indication relates to the general healthy Australian population, the extrapolation of study findings from a diseased study population to the healthy Australian population can be misleading. The relevance of evidence sources that target a population with non-serious disorders or in situations where a continuum of health and disease exists, such as individuals in early disease states, should be considered carefully. In cases where there is data to suggest that the pathophysiology of the disease does not change the way the active ingredient works in the lesser form of disease, compared to a more serious form of disease, the relevance of these evidence sources may be justified.
- It is not appropriate to generalise from studies using defined sub-groups to the general population (e.g. it is not appropriate to use a study on 60–65-year-old adults to support a claim of efficacy in the general population).
- Similarly, if the study was carried out on a mixed sample population, it is not appropriate to claim efficacy in a select sub-group, unless the study specifically addresses efficacy in that sub-group (e.g. if the study was conducted on a mixed sample of adult women and men, it is not appropriate for the indication to relate to pregnant women).
- The results of a study conducted on a homogenous ethnic population group may not be applicable to the general Australian public.
If the target population and the study sample population or sub-group are significantly different, applicants must submit justification accounting for the suitability of the extrapolation (refer to Justifications).
This justification should consider whether extrapolation of the results from the study group to the target population is biologically plausible, as well as relevant environmental and behavioural factors, such as the influence of health practitioner intervention which may differ between populations.
The mechanism of action of the medicine and whether it is applicable to the population/sub-group should be addressed, given that the same results may not be achievable in other populations or sub-groups due to physiological differences.
Consideration should be given to whether the dose requires modification. The justification may use studies on different population groups, non-clinical studies, and in vitro studies to support the pivotal study.
Balance of evidence
Indications must not indirectly, or by implication, lead consumers to believe that the medicine will assist in a health benefit that is not explicitly supported by the balance of evidence i.e. the weight of good quality evidence should agree with the proposed indication. The indication cannot be based on a study that is not consistent with the surrounding body of knowledge (refer to Balance of evidence and conflicting results).
An indication that is consistent with the broader knowledge base and is supported by the balance of evidence is more likely to remain valid for the life of the medicine as new research becomes available.
Presentation
The presentation of therapeutic goods is the way in which the goods are presented for supply, and includes matters relating to the name, labelling and packaging of the goods, and any advertising or other informational material associated with the goods.
For example, aspects of the product that are considered to comprise the ‘presentation’ include:
- the name
- indications
- directions for use
- warning and cautionary statements
- packaging
- dosage form
- logos, symbols and pictures.
Please note that Product Information and Consumer Medicine Information documents may form part of the 'presentation'. These documents are only required for registered medicines and are not required or part of the evaluation of an assessed listed medicine. These should not be provided with an application unless specifically requested.
Unacceptable presentation
The presentation of assessed listed medicines is evaluated pre-market. A product will not be approved if the presentation is deemed to be unacceptable. Unacceptable presentation is defined in subsection 3(5) of the Act and regulation 3A of the Therapeutic Goods Regulations 1990.
The presentation may be unacceptable where the proposed assessed listed medicine is very similar (e.g. in name, packaging, design, colour, flavour and overall presentation) to an existing listed medicine. If the current AUST L with be replaced by the new AUST L(A) medicine, the applicant should provide an assurance that both medicines will not be supplied simultaneously on approval of the AUST L(A) medicine i.e. the new AUST L(A) medicine will not be released for supply to market until supply of the current AUST L has ceased.
If the intention is to co-market the current AUST L with the new AUST L(A) medicine i.e. if the assessed listed medicine will not replace the ‘standard’ listed medicine in the market, then the presentation must be adequately differentiated, (e.g. through the name and label).
For more examples of unacceptable presentations and a detailed outline of many of the considerations for the presentation of medicines, sponsors should refer to Listed medicine presentation and labels. Some considerations for assessed listed medicines are addressed below.
Name
The name of a medicine refers to the identifying descriptor given to the product by the sponsor (e.g. 'Acme Pharmaceuticals Vitamin C Tablets'). For further guidance refer to Labelling medicines to comply with TGO 91 and TGO 92
Assessed listed medicine applications will not be approved if, for example:
- the name of a proposed assessed listed product might be considered as unacceptable presentation
- the name (and any other information on the label) does not comply with the Therapeutic Goods Advertising Code
- the use of a well-known brand name on new products ('umbrella branding') with different active ingredients relative to existing products, for either the same or a different indication, might cause consumers or health care practitioners to confuse existing and new products.
In instances where a brand name may be acceptable, the name of the new product is assessed based on the extent to which it will be immediately apparent to consumers that they are dealing with a different product.
The strength of association of the brand name with the active substance or therapeutic use; the level of differentiation of the presentation of new product relative to current products; and the safety and efficacy in instances where consumers might mistakenly take the wrong product are all considered.
For further guidance, refer to Labelling and advertising non-prescription medicines.
Labels
A label, in relation to therapeutic goods, is a display of printed information on or attached to the medicine; on or attached to a container or primary pack in which the medicine is supplied; or supplied with the container or pack.
Medicine labels should comply with all relevant legislation before the medicine can be supplied in Australia, including advertising requirements. Specific references relating to medicine labelling requirements include:
- The Therapeutic goods labelling order as current and in force
- Part 5-1 (Advertising and generic information) of the Therapeutic Goods Act 1989
- Subsection 3(5) of the Therapeutic Goods Act 1989
- Therapeutic Goods Advertising Code
- Therapeutic Goods Regulations 1990
- Therapeutic Goods (Permissible Ingredients) Determination
- The Poisons Standard
- Naming ingredients.
Additional guidance can be found in:
- Listed medicine presentation and labels
- Labelling and advertising of non-prescription medicines
- Labelling medicines to comply with TGO 91 and TGO 92
- Guidance on applying the Advertising Code rules.
Copies of all draft medicine labels must be submitted with all applications to list new medicines and applications to change the labelling of an assessed listed medicine. A certified English translation of any other language appearing on the label must be provided. All text must be consistent with the English information and not include or imply any additional indications.
During the evaluation, all aspects of the medicine presentation, including proposed labelling and package inserts (if present)19, are assessed for compliance with the various legislative requirements (including advertising requirements). Some specific points are outlined below.
Listing number
Once approved, assessed listed medicines are assigned a unique AUST L(A) number which must be displayed on the label in accordance with the requirements of the Therapeutic Goods Order No. 92 – Standard for labels of non-prescription medicines (TGO 92) and the Therapeutic Goods Regulations 1990.
Ingredients
TGO 92 specifies how active ingredients should be declared on labels.
Excipient ingredients do not need to be on a medicine label, unless required by legislation. For example, Schedule 1 of TGO92 and the Therapeutic Goods (Permissible Ingredients) Determination may require specific labelling associated with certain ingredients.
Where a sponsor chooses to disclose a non-mandatory excipient on a medicine label, then all excipients must be disclosed i.e. declaration of excipients on a label cannot be selective. This may be permitted in some cases, such that selective disclosure of excipients other than those required to be disclosed by the Labelling Order, is justified (e.g. potential consumer safety, cultural or religious preferences) and does not mislead consumers to think they are the only excipients in the medicine.
Reference to a colour, fragrance or flavour (e.g. red capsule, strawberry flavour) is generally acceptable without justification. Sponsors may also choose to include the full formulation on the label.
Statements such as ‘gluten free’ or ‘sugar free’ must be true of all ingredients in the medicine, including proprietary ingredients. The sponsor should provide written assurance in their submission that the product does not include the stated substance.
If the formulation includes a proprietary ingredient, the sponsor should check with the manufacturer or supplier of the proprietary ingredient to ascertain that it does not contain any component it is claimed to be 'free of' on the label.
The sponsor should also check whether the proprietary ingredient contains any specified excipient that must be declared on the labels in accordance with the TGO 92 and the Therapeutic Goods (Permissible Ingredients) Determination.
Any claims relating to excipients will be assessed by the TGA during the evaluation.
Directions for use
Directions for use include the following information for each target population for which the product is intended:
- dosage
- method of administration
- frequency and duration of treatment for each indication
- relevant target population (e.g. age), where applicable.
For liquid products, recommended doses should be able to be measured using commonly available metric measures, or a suitable measure provided in the pack. References to a culinary 'spoonful' (e.g. teaspoon, dessertspoon, tablespoon, etc.) are not acceptable.
Products which are intended for symptomatic relief should include a qualifier such as 'as required' or 'when necessary' after the specific dosage frequency (e.g. 'take one tablet in the morning when necessary'). The directions 'as required' or 'when necessary' are not acceptable on their own.
Certain medicines may require a statement in the directions for use that communicates the duration of treatment to the consumer. This helps manage consumer expectations, prevent confusion, and enable informed decisions by not omitting important information. For example, if the evidence indicates that long-term use is necessary for a health benefit, a statement about the expected length of results may be needed.
Claims
Claims that do not include a specific therapeutic use of the medicine (e.g. ‘contains 30% more’, ‘water resistant’ etc.) are not considered to be indications. Such claims are not required to be in the ARTG entry for a medicine.
Claims that relate to therapeutic use must be consistent with the approved indication of the medicine as it is recorded on the ARTG. The medicine label must not include any claim that is inconsistent with the information included in the ARTG for the medicine and must comply with applicable standards and advertising requirements outlined in Listed medicine presentation and labels and the Therapeutic Goods Advertising Code.
The indications and product labels (including label claims) for assessed listed medicines are pre-market assessed by the TGA for which evidence must be provided at the time of application. Some types of claims have specific requirements, as outlined below.
Claim that product has undergone efficacy assessment
Assessed listed medicines can claim that the product has undergone efficacy assessment. The TGA assessed claim is a symbol and/or statement that indicates that a medicine has had the efficacy for its indications assessed by the TGA. It can only be used in accordance with the TGA's authority. The TGA assessed claim is not mandatory. Refer to Using the 'TGA assessed' claim on medicine labels.
Claims implying a high level of certainty
For claims implying a high level of certainty, including ‘clinically’ and ‘scientifically’ in combination with ‘proven’, ‘tested’, ‘trialled’, ‘evidence based’ etc. refer to Applying the Advertising Code rules and Educating advertisers about compliance with the new advertising code and procedures.
Claims that may lead consumers to believe that the evidence supporting the efficacy of a product is unequivocal are not appropriate unless supported by sound data from robust clinical trials on the product.
Claims of rapid effect
Claims of ‘fast’ or ‘rapid’ action or effect may be used when the indications are for relief of symptoms where the speed of onset is relevant and they are well supported by clinical and pharmacokinetic data. They are not suitable when used in relation to chronic conditions (unless well supported for specific, temporary symptomatic relief such as pain), conditions not requiring immediate relief, or medicines where the pharmacokinetics or mechanism of action precludes a fast action. For further information, refer to Guidelines on presentation aspects of OTC applications.
Warning and disclosure statements
Labels must include advisory statements required by the Therapeutic Goods (Permissible Ingredients) Determination and Poisons Standard. These legislative instruments work in conjunction to regulate ingredients included in all listed medicines, as described in Item 8 of Schedule 4 to the Therapeutic Goods Regulations 1990.
All indications relating to symptoms must be accompanied by the statement ‘If symptoms persist consult your healthcare practitioner’ or words to that effect.
Sponsors should include any relevant warning statements and/or contraindications on the product label. The TGA may also raise the need for relevant statements during the evaluation.
Comparisons and endorsements
Statements comparing a product with other products or treatments must be supported by acceptable evidence and must comply with the Advertising Code.
Claims that a product is endorsed can be made on the label of the product only when such as claim complies with all the requirements of the Advertising Code. For example, if the label states that the product is endorsed by an organisation that represents the interests of healthcare consumers and that organisation has received valuable consideration for the endorsement, that fact must be stated on the label.
Additionally, endorsements must not be given by certain organisations or individuals as specified in ss24(6) of the Advertising Code. All claims, including those relating to endorsements, must be accurate, balanced and not misleading and they must be substantiated by the advertiser prior to being published or disseminated.
Where the endorsement includes a restricted representation, approval for the use of a restricted representation must be granted before it can be advertised (see Advertising – Restricted representations).
References to internet sites or other products
The inclusion of internet addresses on labelling is only acceptable where the information on the website (including any direct links from that website) is not inconsistent with the information included in the ARTG for that product. Websites are considered advertising and are subject to the Advertising Code and should only contain information that is acceptable under other laws, including TGA and ACCC.
Direct references to a sponsor’s other products on the label may be permitted, provided that the products are included in the ARTG and have been pre-market assessed for efficacy. Care should be taken where reference is made to:
- more suitable dosage forms within the same range for different age groups
- another product that can be used in conjunction with the product, where appropriate
- other products within the same product range that have the same trade name as the current product, where appropriate.
Graphics, logos, symbols and market differentiations
Non-corporate graphics, logos or symbols on labels must not be inconsistent with the product's approved information. For example, a graphic highlighting joints would be inappropriate for a product that is indicated for use only on soft tissues.
The presentation of new products should be adequately distinguishable from existing products (see Unacceptable presentation).
Advertising
An advertisement, in relation to a therapeutic good, includes any statement, pictorial representation or design, however made, that is intended, whether directly or indirectly, to promote the use or supply of the goods.
Only product labels for assessed listed medicines are assessed, however all advertisements including all Australian-based websites promoting the supply or use of therapeutic products, must comply with Part 5-1 of the Act and the Therapeutic Goods Advertising Code.
Restricted representations
It is an offence under the Act (see paragraph 42DL(1)(c)) for a sponsor or any other advertiser to use a restricted representation without prior approval of, or permission from, the Secretary of the Department of Health.
To obtain approval to use a restricted representation in relation to the advertising of an existing assessed listed medicine, sponsors may submit an application under section 42DE of the Act by completing the Application for approval to use a restricted representation in advertising form.
If you are proposing to use a restricted representation on your medicine label, please note this in the cover letter of your assessed listed medicine application. Do not submit the Application for approval to use a restricted representation in advertising form with your assessed listed medicine application, as approval for the use of a restricted representation in advertising can only be considered once the assessed listed medicine is listed in the ARTG.
Any application for use of a restricted representation must be consistent with the product's accepted indications or intended purpose, as per its ARTG entry; and/or any mandatory warning or cautionary statements which are required to be included in the product packaging/labelling to satisfy other regulatory requirements.
Application and approval processes
All medicine applications that involve pre-market assessment by the Complementary and Over the Counter Medicines Branch (COMB) follow a similar sequence of processes.
The main difference between applications of different types is that applications in lower categories have shorter assessment timeframes due to the reduction in information to be evaluated.
The steps involved in most applications are:
- pre-submission
- submission
- preliminary assessment
- evaluation and requests for information
- decision
- finalisation
- post-listing.
The sections below provide further information on each of these steps.
Pre-submission
Verifying eligibility and validity
Before submitting an application, potential sponsors should ensure that the proposed product meets the eligibility requirements for the assessed listed medicines pathway.
Checking ingredients
All ingredients (active and excipients) must be included in the latest Therapeutic Goods (Permissible Ingredients) Determination and the formulation must be compliant with any restrictions or requirements associated with those ingredients.
If the product contains any ingredient not specified in the Determination, applicants will need to apply for a new substance for use in listed medicines prior to proceeding. Note that the application for an assessed listed medicine cannot proceed until the ingredient has been successfully evaluated and added to the Determination (see Understanding the application requirements for a new substance in listed medicines). Alternatively, applicants may submit an application for a registered medicine (such as a registered complementary medicine).
If the medicine contains any proprietary ingredients (PIs) - including flavours, fragrances and printing inks - applicants will need the proprietary ingredient ID number. PIs are listed in the Proprietary Ingredients Table. If the PIs are new (i.e. not in the Code tables), applicants must submit the completed Notification of a new proprietary ingredient form to obtain a PI ID number.
All substances in a PI intended for use in listed medicines must be included in the Therapeutic Goods (Permissible Ingredients) Determination. It is the sponsor’s responsibility to confirm that the PI does not contain any ingredients not on the Determination, and that warning statements related to any PI components that need to be declared are included on the label.
Checking the indications and claims
Applicants must ensure that the indications and claims on the label are appropriate for the assessed listed medicines pathway and that they correctly align with the evidence supplied in the application.
This includes ensuring that the formulations, dosage, route of administration, and target populations are all substantially similar to those employed in the studies. If the indications or claims include restricted representations, applicants should note this in the cover letter when submitting the assessed listed medicine application (see Advertising – Restricted representations).
The TGA will evaluate indications that are on the product label(s) and these indications must also be included in the ARTG entry. Any indications referred to in the application dossier should be consistent with the proposed product label(s). Indications should be referred to the same way throughout the application dossier.
Checking evidence
Applicants should check the minimum data requirements for the method by which they intend to support the efficacy claims. For an application to be effective, it must contain the correct type, number and quality of studies; the correct type of pharmacokinetic/ biopharmaceutic studies; and GMP clearance valid for the entire duration of evaluation.
If the GMP clearance is due to expire within the minimum timeframe or is likely to expire before the application is finalised, applicants should either apply to renew the GMP clearance or seek an extension to the GMP clearance expiry before submitting the application.
It is recommended that applicants check all of the relevant European Union and ICH guidelines adopted in Australia for any specific requirements that may apply.
Label proofs
Proofs of the proposed labels must be submitted. The labels should comply with the relevant Therapeutic Goods Orders and must include at least one intermediate level indication. All indications and claims on the label will be assessed by the TGA. Only assessed indications will be included in the ARTG entry for the product if it is approved.
Determining the application category and compiling the dossier
It is important that applicants determine the application category correctly. If the application does not meet the requirements of the selected category, and does not include the required data, it will not be accepted for evaluation. Refer to Application categories for assessed listed medicines.
Pre-submission meeting
Applicants may wish to request a meeting with the TGA prior to applying for a new assessed listed medicine. See pre-submission meeting with the TGA for details on arranging a meeting. There is no fee associated with a pre-submission meeting. The TGA may be able to address any questions proposed for the pre-submission meeting in writing in which case a pre-submission meeting will not be required.
Pre-submission meetings provide an opportunity for applicants to seek clarification of the requirements and to revise the approach to their application. These meetings may help applicants submit a high quality and complete dossier and consequently ensure that the evaluation process proceeds smoothly and in line with legislated timeframes.
During the pre-submission meeting, discussion will focus on the structure of the proposed application, the identification of critical issues and the suitability of the proposed approach. The TGA does not assess or evaluate the application as part of a pre-submission meeting.
After the meeting, the applicant forwards a meeting record to the TGA and any other participants. The meeting record is intended to be a summary that clarifies the agreed outcomes and any actions arising; it is not intended to be a transcript. The TGA will acknowledge the meeting record. If necessary, the final meeting record should be included in Module 1 of the application dossier.
To arrange a free meeting with the TGA, follow the general guidance on pre-submission meetings.
Application submission
Completing and submitting the application
The TGA Business services portal provides an electronic facility for the listing and registration of medicines on the ARTG. Applications are created and lodged through TGA Business Services. Applicants will need a Client ID number and a password access to the TGA Business services. Applicants who do not have a Client ID number or access to TGA business services should submit the online organisation details form.
The Listed and assessed listed medicines: application and submission user guide provides instructions on how to create and submit your assessed listed medicine application.
Once the application has been submitted, applicants will be issued with a unique submission number that can be used in all future communications about the application.
Withdrawing an application
An application can be withdrawn at any time up until the decision is made. This can be done through the Business Services. Alternatively, applicants can advise the TGA in writing of the intention to withdraw the application.
When an application is withdrawn, the TGA may retain the application and any material submitted in connection with the application.
Application fee payment
Once an application has been received by the TGA, an invoice will be issued for the application fee. For details of the current fees, refer to Summary of fees and charges to applications submitted to the TGA.
For information on fees and the available payment methods see:
Preliminary assessment
The TGA will conduct a preliminary assessment of the application to determine whether it meets the requirements to proceed to evaluation. This is simply a quality assurance process, and no evaluation of the content of the application is undertaken at this point.
Assessed listed medicines have two fees: an application fee (for preliminary assessment) and an evaluation fee (payable if the application passes preliminary assessment and proceeds to the evaluation phase).
Applications that pass preliminary assessment
The application will pass preliminary assessment if it meets the requirements under section 23B of the Act. Generally, this means that:
- the prescribed application fee has been paid
- the application includes all required information (i.e. in adherence to the Mandatory requirements for an assessed listed medicine application, CTD Module 1: Administrative information for assessed listed medicines and General dossier requirements), for the correct application category, to enable the TGA to evaluate the application.
Applicants will have an opportunity to make minor corrections detected during the preliminary assessment process if the issue can be rectified promptly. For example, if the evaluators cannot locate an attachment mentioned in the application, the TGA will provide an opportunity to submit the attachment.
If the application passes preliminary assessment, the applicant will be notified in writing that the application has been accepted for evaluation. The evaluation process will not commence until the evaluation fee has been paid in full.
Applications that do not pass preliminary assessment
If the application does not pass preliminary assessment, it will not be accepted for evaluation or required to pay the evaluation fee. The applicant will be notified in writing and an explanation of why the application was not effective will be provided. The application fee will not be refunded. Applicants are not able to appeal this decision under section 60 of the Act.
Lapsing applications
The application will lapse if evaluation fees are not paid within 28 days of becoming payable. The TGA will notify the applicant of the lapsing of the application. A new application must be submitted, and a new application fee paid, if the application lapses.
Evaluation and requests for information
Once an application has passed preliminary assessment and the evaluation fee has been paid, the TGA will evaluate the application.
Evaluation
The TGA assesses the efficacy data and the product label, which includes:
- efficacy evidence (a review of the types, quantity, quality and validity of studies)
- the proposed indication(s) and label claim(s)
- relevant parts of the medicine presentation for compliance with legislative requirements (including labelling and advertising).
Requests for information (RFIs)
The TGA may make a request under section 31 of the Act for additional information to clarify or address issues identified during evaluation. During this time, the evaluation clock will stop. However, evaluators may also seek clarification of minor issues on an informal basis. The clock will not stop in these circumstances.
The applicant should provide an electronic copy of the requested information. No additional unsolicited data will be accepted.
Applicants will be notified of the timeframe for the response. It is important that applicants respond to the request within the timeframe provided. Although the TGA may grant extensions to the due date, this will only be done at the discretion of the delegate if the request is received well before the due date, and if the applicant provides a reasonable justification as to why the extension is necessary.
If the response is not received within the timeframe specified, or if the issues identified in the request remain unaddressed, the application will proceed to the decision phase without the additional information. This may result in the refusal of the application.
The time between the request being issued and receipt by the TGA of the applicant’s response will not be counted as part of the evaluation timeframe (i.e. the clock will stop).
Expert advisory committee advice
The TGA may decide to seek advice from an expert advisory committee, such as the Advisory Committee on Complementary Medicines (ACCM). The TGA will notify applicants of the date of the committee meeting and provide an opportunity for a submission for the committee’s consideration. Any relevant advice received from the committee will be communicated to the applicant.
The decision
When making the decision under section 26AE of the Act on whether to list the medicine in the ARTG, the decision maker (the delegate of the Secretary of the Department of Health) will review relevant documentation associated with the application, including the dossier, evaluation reports, responses to requests for information, and advice from expert advisory committees.
Decision to list the medicine
If the delegate considers the efficacy of the medicine has been established and makes a decision to list the medicine, the TGA will notify the applicant in writing of the decision.
The decision letter will outline standard and specific conditions on the listing of the medicine under section 28 of the Act. The standard conditions that will automatically apply to the listing in ARTG are set out in Therapeutic Goods (Listed Medicines - Conditions of Listing) Determination 2022.
It is important that applicants read, understand and comply with these conditions. If the sponsor does not comply with any one of these conditions of listing, the medicine may be cancelled from the ARTG.
The decision letter will request that the sponsor provides assurance that all details of the medicine are correct before the ARTG entry is created.
Decision not to list the medicine
If the decision is not to list the medicine, the decision letter will include a statement of the reasons for the decision and information on the applicant’s rights to seek a review of the decision. Applicants can appeal this decision under section 60 of the Act. Further see Requesting the Minister for Health to reconsider our initial decision.
Finalisation
Patent certification under the Australia / USA free trade agreement
Sponsors need to provide a patent certificate under subsection 26B(1) of the Act, or notification that this is not required before the medicine can be listed in the ARTG.
If this was not provided with the application, one of the following documents should be completed and submitted via email to nonprescriptionmedicines@health.gov.au, quoting the submission number:
- approved form for notification that 26B(1) certificate is not required
- approved subsection 26B(1)(a) or (b) certificate.
Listing the medicine
Once the completed and signed notification form or patent certificate has been received, the TGA will list the medicine in the ARTG and the product will receive a unique AUST L(A) number.
Sponsors can download the certificate of listing from the TGA Business Services. To do this, follow the guidance on printing an ARTG certificate. The listing of the medicine will commence on the day specified in the certificate of listing. The medicine cannot be lawfully imported, exported, or supplied prior to this date.
The product details will usually be viewable on the TGA Business Services website the day after the information has been recorded in the ARTG.
Post-listing
Publication of outcomes
The TGA may choose to publish a notification of the approval of the medicine on the TGA website. This notification may include the name of the medicine, the sponsor, the approved indications, and any other supporting information necessary. No confidential information (e.g. trade secrets) will be published.
Notices of restricted representations approved under section 42DF and 42DK of the Act are published on the TGA website.
The TGA will also publish a List of assessed listed medicines with data protection.
Annual charges
Annual charges will apply to all medicines included in the ARTG.
The charge is applied annually in July of each year for existing entries in the ARTG, or upon listing of the goods in the ARTG during a financial year. The annual charges apply to any product in the ARTG at any time during a financial year, regardless of whether the product is subsequently cancelled within the same financial year.
Any new product entering the ARTG will qualify for an annual charge exemption (ACE) until such time as the product generates turnover (refer to Annual Charge Exemption Scheme).
Post-market compliance
Assessed listed medicines, like all listed medicines, may be selected for a post-market compliance review at any time. The TGA will check the medicine’s compliance against the regulatory requirements that are self-certified by the sponsor. Efficacy of the product will not be routinely reviewed post-market. For more information refer to Listed medicine compliance reviews.
Timeframes and fees
Timeframes
There are statutory evaluation timeframes for the pre-market evaluation of assessed listed medicines. These are provided below.
| Application category | Notification of preliminary assessment | Evaluation timeframe |
|---|---|---|
| L(A)1 | 40 | 45 |
| L(A)2 | 40 | 60 |
| L(A)3 | 40 | 150 |
See the Changing a listed or assessed listed medicine in the Australian Register of Therapeutic Goods (ARTG) for the timeframes for change application categories.
The above evaluation timeframes:
- apply to working days only, and exclude public holidays and weekends
- exclude where the evaluation clock has stopped e.g.
- for the time taken by the applicant to provide responses to formal requests for information made by the Secretary under section 31
- when the applicant and TGA agree to a mutual stop clock
- only commence once an application has been accepted for evaluation and following payment of the evaluation fee. The delegate of the Secretary will notify an applicant in writing within 40 working days of receiving the application whether the application has passed preliminary assessment.
Although the TGA is required to complete the evaluation within the specified timeframes, the Commonwealth and its representatives are not liable to a person for loss, damage or injury of any kind that is caused by or arises from a failure to decide an application within the specified timeframe.
Fees
The fees are published on the TGA website. There is an application fee for preliminary assessment, and an evaluation fee.
- the application fee cannot be waived, reduced or refunded
- if the application is withdrawn before it enters the evaluation step in the process, the evaluation fee will be refunded. The application fee will not be refunded
- in exceptional circumstances, the Secretary may waive or reduce an evaluation fee if there is information to enable an evaluation to be abridged.
Appendix A Considerations of evidence quality in efficacy evaluation
Study scope
Population selection
The study population used should be appropriate for the outcomes being tested (e.g. healthy individuals, women, elderly, adults etc.). Data obtained from studies using participants with serious health concerns is generally not appropriate to support an indication for assessed listed medicines; unless the indication relates directly to a population with that condition (e.g. a restricted representation). In some circumstances it is also possible to use studies of participants with serious health concerns where positive outcomes were noted, to provide secondary (non-pivotal) sources of evidence.
The study population should be clearly identified in the study protocol, and all the inclusion and exclusion criteria adequately outlined. These criteria are particularly important. If the criteria are too lax, the validity of the inclusion may be questionable, while if the criteria are too tight, the results may not be applicable to the wider population (refer to External validity).
Baseline characteristics of treatment and control groups should be documented to ensure equivalence in key areas such as age, weight, diet and other factors that may contribute to non-treatment differences in health benefit between groups.
Sample size
Studies submitted in support of the efficacy should involve enough participants to enable the reliable detection of clinically significant treatment effects. The number of participants required to be reasonably certain of a reliable result is described as the ‘power’ of a study. It is the likelihood of the study finding a true difference between treatments if one exists. The larger the sample size, the greater the statistical power i.e. the greater the power to detect a smaller difference.
Applicants should carefully consider any limitations of the statistical calculations that the study authors have reported, including the number of drop outs and the impact this may have on the reported study outcomes.
To ensure that a study can support the indications for a product, the power should be at least 80 %. The study should also provide a description of how the power/ sample size was determined. Underpowered studies may be submitted as supplementary evidence, however cannot be used as primary evidence due to the statistical uncertainty of the effect.
Further discussion on sample size can be found in the EMA ICH E9 statistical principles for clinical trials - Scientific guideline.
Studies submitted to support indications should have a statistical power of at least 80% to ensure robust and reliable results.
Study endpoint(s)
The primary variable/outcome/endpoint of a study is the measurement that provides an estimate of the effect or outcome of a treatment. In general, studies should only have one primary endpoint that:
- is the most clinically relevant to the proposed indication, and consequently, capable of demonstrating the efficacy of the medicine
- provides a reliable and validated measure(s) consistent with the standards and norms of the relevant field
- is clearly defined in the protocol before the start of the study. This is to avoid artificial result selection via post-hoc definition of the endpoint.
If possible, multiple measures for endpoints should be used.
Secondary endpoints assess other effects of the medicine that may or may not be related to the primary endpoint (e.g. questionnaires to assess subjective pain, pain assessed at two different timepoints, or pain and quality of life assessed at the same timepoint).
If a study includes secondary efficacy endpoints, they should provide supportive evidence for the primary endpoint and needs to be pre-specified and part of an appropriately planned statistical analysis strategy.
Secondary endpoints that are statistically significant and clinically relevant can only be used after the primary objective of the clinical trial has been achieved. When the primary endpoint has not been achieved, the results of the secondary endpoints are considered exploratory and should not be used to support efficacy.
An endpoint may have composite variables and such variables could only be considered to support an indication if it was pre-specified and embedded in a valid confirmatory analysis strategy. The EMA guideline Points to consider on multiplicity issues in clinical trials provides further explanation on handling composite variables and its components.
In some instances, the direct measurement of a clinically relevant benefit will be neither feasible nor practical (e.g. for the demonstration of a long-term health benefit). In such circumstances a surrogate variable, which relates to a clinically important outcome but does not in itself measure a clinical benefit, may be used. The surrogate variable must be a demonstrably valid and reliable predictor of clinical benefit.
In all cases, applicants should provide a justification addressing the clinical relevance of the outcome and the rationale for its selection. Additionally, they should demonstrate that the methods used to measure the variables that contribute to the study endpoints are validated and meet appropriate standards for accuracy, precision, reliability, reproducibility, and responsiveness. Results for every measured outcome must be reported, regardless of whether they are positive, negative, or non-significant. The report should also report and discuss any side effects or adverse events observed.
Internal validity
TGA evaluators review the extent to which the design of the study eliminates sources of bias, and therefore, provides confidence in interpreting the results.
Some of the important considerations for assessing the internal validity of evidence are outlined in the sections below. They are, however, dealt with in greater detail in TGA adopted guidelines. Other online references with tools and checklists for appraising the quality of studies include CASP, CEBM, JBI’s Critical Appraisal Tools, and Risk of bias tools.
Sources of bias
The ideal experimental design should compare two groups that do not differ in any significant respect apart from the treatment or intervention of interest. Significant differences between the groups may introduce bias into the comparison. Bias is the tendency of any factors associated with the design, conduct, analysis and interpretation of the results of a study to cause the treatment effect deviate from its true value. Bias can be introduced through deviations in conduct (operational bias) or may be inherent in the design and analysis of the study (statistical bias).
There are a number of ways to assess risk of bias for different studies (refer to NHMRC assessing risk of bias guideline, Table 1) with the most commonly employed tool for assessing risk of bias for randomised clinical trials being the Cochrane Risk of Bias tool. A study should adequately control for bias and consider the impact of identified biases on the reliability of the study.
Control, randomisation and blinding
Many sources of bias can be addressed using appropriate controls, randomisation, outcome selection, and blinding of both researchers and patients.
Applicants should consider the controls, randomisation, blinding, and allocation concealment (where appropriate) when assessing the reliability of their evidence as support for indications. Well-designed and conducted studies should include a detailed description of the subject eligibility criteria, method of randomisation, and blinding technique employed to enable assessment of the potential for error or unblinding. Studies that do not employ or report these bias reduction techniques are less likely to be accepted by evaluators as convincing evidence of the veracity of claims. The TGA-adopted EMA Note for guidance on choice of control group in clinical trials has further information on general principles in choosing a control group.
Conduct
All studies should be conducted according to Good Clinical Practice (GCP) principles and have appropriate ethical certification. They should also be compliant with International Council for Harmonisation (ICH) and European Medicines Agency (EMA) guidelines adopted by the TGA. These can be accessed on the TGA website.
Clinical trial protocol
The clinical trial protocol is key in the planning, conduct, interpretation, monitoring, analysis and reporting of a clinical trial. A well-written protocol provides consistency and ensures rigour of trial conduct and allows for a full appraisal of the results after trial completion. The EMA Guideline for good clinical practice E6(R2) outlines the contents that should be included in a clinical trial protocol. The SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) 2013 can be used as a guideline for the minimum content of a clinical trial protocol.
Departures from the planned conduct of a study may introduce operational bias. It is therefore important that any evaluation of a clinical study report consider the number and severity of protocol violations and deviations and the completeness of patient follow-up. These may include changes to the inclusion or exclusion criteria, outcomes measured, group sizes, treatment protocol, blinding, and/or duration. Any statistical consequences of these changes and revision of analytical approaches should also be addressed.
Reporting
Clinical study reports should be adequately documented following the approaches outlined in other ICH guidelines (see ICH E3 and E6).
The EQUATOR Network provides reporting guidelines for all main study types and is recommended to assist in reporting trial outcomes. There are extensions to the guidelines that are relevant to complementary medicines (e.g. herbal medicines and Chinese herbal medicines) that may be useful as reference.
Dealing with missing data
One of the problems with clinical studies, particularly longer-term studies, is that planned data points may be missing due to subject attrition (patient dropouts, treatment failures, non-compliance, adverse events etc.) and missed measurements, among other factors.
Missing data stemming from high attrition rates or missed measurements can lead to non-comparability of treatment and control groups, a reduction in the statistical power of the study, and the introduction of significant bias. This may make the results of the study difficult to interpret and diminish the ability of the study to support an indication or claim. Consequently, the design and conduct of a clinical trial should seek to minimise the amount and/or impact of missing data and where relevant. The TGA-adopted EMA Guideline on missing data in confirmatory clinical trials provides further guidance on dealing with missing data.
There is no absolute threshold for the proportion of missing data that would mean a trial was not suitable to support any specific indication or claim. The TGA will consider the reasons for the missing data, whether appropriate imputation methods were used, and the likely effect of any missing data on the study results.
Statistical validity
Statistical validity refers to the extent to which a measurement is well-founded and accurately reflects reality. The validity of the conclusions of the study is critically dependent on the nature of the metric used to measure an effect, the size of effect, and the type of statistical transformations and analyses performed.
Studies must use valid statistical methods to assess the outcomes and must account for any potential confounders. These statistical methods should be fully described in the protocol and should be appropriate for the efficacy outcomes measured. Unplanned analyses undertaken after the completion of a trial (post-hoc analyses) are to be avoided as they are unlikely to have been considered in power calculations and study design.
The principles outlined in Note for Guidance on Statistical Principles for Clinical Trials (ICH Topic E9) is a good resource when assessing whether the statistical analysis of a clinical study has been conducted in a robust manner.
Analysis set
The set of subjects whose data are to be included in the main analyses should be defined in the statistical section of the protocol.
The intention-to-treat (ITT) analysis includes all randomised subjects according to the allocated treatment regimen, rather than the actual treatment experienced (i.e. subjects are analysed according to their allocation, regardless of their compliance to the treatment). This is the most conservative approach as it biases the analysis towards the null hypothesis.
The other analysis set is a per-protocol (PP) analysis, which is an analysis on the subset of study subjects that complied with the protocol sufficiently. While it is less conservative than ITT analysis, it maximises the chances of an effect being observed and is heavily prone to bias as adherence to the protocol may be related to the treatment outcome. PP analysis is different from complete case analysis where the analysis only includes participants with no missing data on the variables of interest.
When an ITT is performed, all efforts should be made to obtain outcome measurements from all original participants at the end of the study where possible. When data is missing for a participant, imputations methods for missing data may be required (refer to Guideline on missing data in confirmatory clinical trials). Ideally, any missing data in PP analysis will need to be imputed as appropriate.
The choice of using an ITT analysis or PP analysis is dependent on the study design, objectives, research question, and the expected treatment effect. It may be appropriate to perform both analyses and conduct a sensitivity analysis to examine the consistency in results and robustness of data. Decisions concerning the analysis set should be guided by the following principles: (i) minimising bias, and (ii) avoiding inflation of type I error.
Further information on analysis sets can be found in Note for Guidance on Statistical Principles for Clinical Trials (ICH Topic E9).
Statistical significance
Even if the study is well-conducted and sources of bias are limited, there is a possibility that the results arose purely by chance. It is therefore essential that studies use appropriate statistical methods to minimise Type I errors. A Type I error (false positive) is a conclusion that there is a difference between two treatments when no difference exists in reality.
Well-conducted studies will usually report the degree of statistical significance (p-value) associated with the observed difference between treatments. The p-value provides an indication of the probability of claiming that there is a treatment effect when in fact there is no real effect (i.e. the probability of making a Type I error).
The p-value provides an indication of whether the treatment effect that has been observed can be explained by chance alone. Although there is no definitive p-value threshold, the lower the p-value the greater the likelihood that the effect observed is real. In practice, a ‘p’ value of less than 0.05 indicates with acceptable certainty that an observed effect or health benefit is unlikely to be due to chance.
In considering the strength of the evidence, applicants should ensure that:
- the statistical test used to derive the p-value is appropriate and reliable
- the p-value obtained for the primary endpoint is less than 0.05
- all the actual p-values (not just p < 0.05) are reported.
It is possible for the significance threshold to differ from p < 0.05, however, the choice of p-value needs to be pre-specified and justified based on the research question being asked. It is important to bear in mind that statistical significance does not provide information about the magnitude of benefit produced or whether it is likely to be clinically meaningful.
Confidence interval
The confidence interval (CI) is the range of values within which there is a certain likelihood that the true value can be found. The confidence level is the probability that the CI contains the true difference. CIs can range from 90 % to 99 % though well-conducted studies should usually report the 95% CI. This means that there is a 95 % chance that repeated experiments would have outcomes that fall within the specified range.
When comparing treatment groups, 95 % CIs help with assessing the precision and range of the estimated treatment effect. A wide CI indicates that there is less confidence that the effect estimate reflects the true population effect (see Figure 1). As such, the confidence in the evidence may be considered lower because of resulting uncertainty about the results. Causes of a wide CI around the estimated effect include small sample sizes, low event rates, outliers in the data, high variance, or higher confidence level.
Clinical significance
A statistically significant outcome indicates that there is likely to be a relationship between intervention and outcome. For a study to provide adequate support for an indication, the observed differences should be statistically significant and clinically meaningful.
Clinical significance is a degree of benefit that is considered worthwhile to justify a particular treatment or intervention. It can be regarded as a measure of how meaningful a particular study outcome might be to the consumer.
For example, a study might demonstrate a statistically significant weight loss, but in practice, that effect may not generally be experienced or noticed by members of the wider population.
However, defining and quantifying the true ‘significance’ of study outcomes is not always straightforward. Divergent views on what is clinically significant, such as individual relevance and treatment goals, and differences in risk-benefit views may exist between patients and/or between patients and clinicians.
Clinical significance is more readily demonstrated when dysfunction is significant, noting that the health outcomes provided by assessed listed medicines may be modest, for less serious conditions, not readily apparent, and/or achieved over long periods of time.
Judgements about clinical significance are often made by experienced clinicians within a context of ongoing monitoring and supervised care and patient experience. Given the meaningfulness of a predetermined ‘significant clinical benefit’ may vary between patients depending on a number of factors such as state of disease, comorbidities, personal circumstances, and alternative options for treatment, it can be challenging to determine to the practical health outcomes in a study.
Applicants should consider the meaningfulness and benefit of an observed health outcome to the intended target population. In making such judgements, it is useful to consider the significance, confidence interval, confidence level, and the magnitude of the treatment effect.
Clinically significant changes in outcomes can be determined by a predetermined threshold tested in clinical studies and are identified interchangeably by terms such as ‘minimal clinically important differences (MCID)’, ‘clinically important differences (CID)’, and ‘minimally important changes (MIC)’.
These thresholds need to be applied with discretion as there are no standards for calculating clinically important changes in outcomes. Determining clinical significance threshold is subjective due to variations in patient status and goals and clinician experience. The different interpretations of clinical study outcomes are illustrated in Figure 1.
Figure 1: Illustration of the impact of the size of the effect and the precision of the 95% CI on the interpretation of the outcomes of a clinical study.
{kind=link}
A forest plot chart illustrating different levels of statistical and clinical significance in research outcomes.
The plot consists of 6 horizontal lines representing different scenarios:
- "Statistically and clinically significant": Shows a confidence interval entirely to the right of the "Clinically significant" line.
- "Statistically but not necessarily clinically significant": The confidence interval crosses the "Clinically significant" line.
- "Statistically but not clinically significant": The confidence interval is between "No difference" and "Clinically significant" lines.
- "Not statistically significant and inconclusive clinical significance": A wide confidence interval crossing both the "No difference" and "Clinically significant" lines.
- "Neither statistically nor clinically significant": A narrow confidence interval close to the "No difference" line.
- "Statistically significant reversed effect": The confidence interval is entirely to the left of the "No difference" line.
Each scenario is labelled with its p-value (either p<0.05 or p>0.05) on the right side. The x-axis shows "No difference" and "Clinically significant" threshold markers.
This visualisation helps interpret research results in terms of both statistical and clinical significance.
When there are no specific guidelines on what magnitude of effect is considered clinically significant, it is worth considering the size of the CI for the difference, whether it covers the pre-defined target outcome, what proportion of the CI is in the region of interest and what proportion of patients achieved the outcome pre-specified in the protocol. When the clinically significant difference is unknown, it is reasonable to apply the treatment effect size used to calculate the sample size.
Ancillary endpoints that corroborate the clinical outcome can also be used when there is no agreed standard for MCIDs. For example, ancillary endpoints for pain outcome measures include lower rate of analgesic use in treatment vs. control (in terms of frequency, strength, and duration of benefit), which quality of life domains improved and frequency of visits to medical professionals for pain management.
There are many approaches to assessing clinical significance and it is important to determine the approach a priori and to identify the expected clinical benefit for the patient.
External validity
External validity and extrapolation are important considerations in determining the scope of an indication.
Extrapolation is the application of results from a study to a different population from the one used in the study (e.g. results from a study on 20-25 year old women being applied to 30-35 year old women). Generalisability (or external validity) is a term often used to conceptualise the extent to which study results can be broadly generalised beyond the setting of the study and the particular sample groups used.
The validity of such inferences depends on the representativeness, size, and variability of the study sample. The greater the extent of these characteristics, the more generalisable the results. Additional factors to consider when determining if the results of a particular study can be extrapolated or generalised are:
- The effect of gender, age, or ethnicity. Are physiological differences likely to impact on the efficacy of the treatment?
- The timing of the treatment. The stage of the condition /illness may impact on treatment outcomes.
- Variations in the condition being treated. There may be distinct underlying aetiologies despite similar presentations.
It is also worthwhile considering whether the results of a particular study are applicable to individuals as well as groups. Applicants considering extrapolation of the results from the study population to the target population should give consideration whether this is biologically plausible, and if the target population would respond to the intervention in a similar way to the study population.
Balance of evidence and conflicting results
The strength of evidence provided by a specific study is greatly enhanced if the effect is reproducible, and if the cause-effect relationship proposed is consistent with existing knowledge.
A well-conducted literature search will identify all related studies, and these should be assessed in relation to the findings of the primary evidence. Positive, null and negative results should be examined. If there are conflicts in the outcomes of different studies, the applicant must provide a plausible explanation for the conflicts in a scientific justification.
In some instances, the conflicts can be readily accounted for by differences in design or methodology (e.g. dose form, population, timing etc.). If suitable explanations for the discrepancies cannot be found, the highest quality studies will receive higher weight in the evaluation.
Summary of considerations for studies
Table 9 summarises many of the considerations outlined in the preceding sections. Applicants may find it helpful in appraising the quality of their evidence prior to submission of an application.
These principles reflect issues that are taken into account in an ideal study. If there are valid scientific reasons for a particular omission or deviation, applicants can submit a scientific justification which addresses the matter (refer to Justifications).
| Topic | Consideration | Principle |
|---|---|---|
| Scope | Sample |
|
| Outcomes |
| |
| Quality/Internal validity | Bias |
|
| Conduct |
| |
| Changes and missing data |
| |
| Statistics | Basis |
|
| Analysis |
| |
| Significance |
| |
| Balance of evidence |
|
Footnotes
- Medicines listed in the ARTG are authorised for supply in Australia and permitted for export (provided the medicine is unchanged to the one authorised for supply in Australia). Further guidance can be found in Exporting medicines from Australia.
- The therapeutic use relevant to assessed listed medicines are outlined in Schedule 4(8)(d) of the Therapeutic Goods Regulations 1990: (d) the indications proposed by the sponsor of the medicine are either:
- uses of the medicine in preventing, curing or alleviating a disease, ailment, defect or injury in persons, other than a form of the disease, ailment, defect or injury that, under the Therapeutic Goods Advertising Code, is a serious form; or
- uses of the medicine in connection with alleviating a disease, ailment, defect or injury in persons, being a form of the disease, ailment, defect or injury that, under the Therapeutic Goods Advertising Code, is a serious form;
- The Therapeutic Goods Act 1989 defines label as a display of printed information on or attached to the goods, container or primary pack, or supplied with the container or pack.
- A restricted representation is any reference expressly or by implication, to a serious form of a disease, condition, ailment or defect identified in section 28 of the Therapeutics Goods Advertising Code. It is generally accepted to be beyond the ability of the average consumer to diagnose, evaluate, and/or safely treat without consulting a suitably qualified health care professional, however, excludes conditions where the serious form has been medically diagnosed and medically accepted as being suitable for self-treatment and management. Prior TGA approval is required before an advertisement may refer to a restricted representation.
- A serious form is defined in the Therapeutic Goods Advertising Code if:
- it is medically accepted that the form requires diagnosis or treatment or supervision by a health practitioner who is suitably qualified, except where the form has been medically diagnosed and medically accepted as being suitable for self-treatment and management; or
- there is a diagnostic (including screening), preventative, monitoring, susceptibility or pre-disposition test available for the form (including a self-administered test), which requires medical interpretation or follow-up.
- ‘Cure’ is considered to imply complete resolution.
- 'Serious' is defined in the Therapeutic Goods Advertising Code as a form of a disease, condition, ailment or defect which require a suitably qualified health professional to diagnose and/or treat them. This excludes conditions that are suitable for self-treatment once medically diagnosed.
- Sometimes referred to as the 'innovator', ‘originator’ or ‘parent’ medicine.
- As defined in the Therapeutic Goods Regulation 1990, Regulation 2.
- ‘same safety and efficacy properties’ includes, but is not necessarily limited to, the indications and directions for use.
- A biowaiver is an acknowledgement that in vivo bioavailability and/or bioequivalence studies may be considered unnecessary for product approval.
- A study or justification may be required if there is doubt as to whether absorption occurs.
- For example, additional biopharmaceutic and pharmacokinetic studies may be required for non-conventional dosage forms such as modified release products.
- See guidance on preparing the literature search report.
- The finished product is the final dosage form with all active and excipient ingredients.
- Up to 2 % of the total formulation can change compared to the formulation in the evidence, if the change is only to a flavour, fragrance and/or colour (including printing inks).
- All indications for assessed listed medicines must be supported by scientific evidence of efficacy, however traditional evidence is required to support use in a traditional context.
- Although this guidance refers to prescription medicines, the same principles apply for providing biopharmaceutic studies for the relevant assessed listed medicines application types.
Page history
Title changed from 'Assessed listed medicines evidence guidelines' to 'Understanding the application requirements for an assessed listed medicine' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
- Updated contact email address
Description of changes (v1.3)
- Updated link for Oxford Centre for Evidence-Based Medicine: Levels of Evidence (page 28).
Description of changes (v1.2)
- Updated information relating to Therapeutic Goods Orders, Comparable Overseas Body process, Changing a listed or assessed listed medicines, Data Protection Scheme, Advertising Code, restricted representation approval process, TGA assessed claim, and pre-submission meeting guidance.
- Added links to relevant guidance on literature search report, bioequivalence, and evidence requirements.
- Added information on presenting indications on label and guidance on unacceptable presentation when a current AUST L is to be replaced by the new AUST L(A) medicine.
- Clarified indication risk classifications and guidance for structuring an indication.
- Clarified standards of evidence for evaluating efficacy and relocated from the main body to Appendix A.
- Other minor amendments such as formatting and typographical changes, and updates to hyperlinks.
Description of changes (v1.1)
- Modified evidence for low level indications, consistent with listed medicines evidence requirements.
- Removed cross-references to guidelines to avoid confusion. Additional information on biopharmaceutic studies and biowaivers.
- Clarified use of CORs.
- Included cross-reference to ‘Guidance for completing the application form for an assessed listed medicine’.
- Amended pre-submission meeting process.
- Corrected appeal of preliminary assessment decisions and timeframe for lapsing applications. Other minor amendments.
Original publication (v1.0), authored by Complementary and OTC Medicines Branch, TGA.
Title changed from 'Assessed listed medicines evidence guidelines' to 'Understanding the application requirements for an assessed listed medicine' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
- Updated contact email address
Description of changes (v1.3)
- Updated link for Oxford Centre for Evidence-Based Medicine: Levels of Evidence (page 28).
Description of changes (v1.2)
- Updated information relating to Therapeutic Goods Orders, Comparable Overseas Body process, Changing a listed or assessed listed medicines, Data Protection Scheme, Advertising Code, restricted representation approval process, TGA assessed claim, and pre-submission meeting guidance.
- Added links to relevant guidance on literature search report, bioequivalence, and evidence requirements.
- Added information on presenting indications on label and guidance on unacceptable presentation when a current AUST L is to be replaced by the new AUST L(A) medicine.
- Clarified indication risk classifications and guidance for structuring an indication.
- Clarified standards of evidence for evaluating efficacy and relocated from the main body to Appendix A.
- Other minor amendments such as formatting and typographical changes, and updates to hyperlinks.
Description of changes (v1.1)
- Modified evidence for low level indications, consistent with listed medicines evidence requirements.
- Removed cross-references to guidelines to avoid confusion. Additional information on biopharmaceutic studies and biowaivers.
- Clarified use of CORs.
- Included cross-reference to ‘Guidance for completing the application form for an assessed listed medicine’.
- Amended pre-submission meeting process.
- Corrected appeal of preliminary assessment decisions and timeframe for lapsing applications. Other minor amendments.
Original publication (v1.0), authored by Complementary and OTC Medicines Branch, TGA.