UDI requirements for specific device types
Learn about UDI requirements for individual device types, such as implantable devices, software, and surgical loan kits.
Generally, all devices in scope of Unique Device Identification (UDI) requirements must meet all UDI requirements for UDI allocation, UDI labelling and packaging, and UDI record creation and maintenance.
However, some devices are exempt from certain requirements or have additional requirements due to the nature of the device. It is important to understand if the devices that you supply have any specific requirements.
Please note that if the device is of a device class that is not in scope of UDI, it does not need to meet UDI requirements.
For example, reusable devices must meet UDI requirements. However, Class I devices are not in scope of UDI. Therefore, Class I reusable devices do not need to meet UDI requirements. However, Class Is, IIa, IIb and III reusable devices must meet UDI requirements.
Reusable devices
Reusable devices must meet UDI requirements.
Reusable devices must also have the UDI Carrier directly marked onto the device itself if they are intended to:
- be used more than once on different patients
- undergo high-level disinfection or sterilisation before each use.
As a manufacturer, you are responsible for determining if your reusable devices must be directly marked.
The UDI Carrier directly marked onto the device itself must be:
- readable after each reprocessing
- able to withstand reprocessing for the lifetime of the device.
Tash the manufacturer
Tash manufactures reusable scalpels. Because Tash’s devices are intended to be reprocessed and used on another patient, Tash must directly mark her scalpels with a UDI.Tash also manufactures single use syringes. As the syringes are not reusable, Tash is not required to directly mark her syringes with the UDI. Tash must still meet all other labelling requirements for her single use syringes.
Reusable devices exempt from direct marking requirements
Reusable devices do not need to be directly marked if they are:
- intended to be used on one patient only and no subsequent patients
- not intended to undergo high-level disinfection or sterilisation before each use
- devices principally sold in retail premises.
Additionally, reusable devices are exempt from direct marking where:
- any type of direct marking would interfere with the safety or performance or effectiveness of the device
- it is not technically feasible to directly mark the device
- the device was manufactured and labelled prior to the direct marking compliance date.
For more information on direct marking, see Complying with the Unique Device Identification requirements for medical devices.
Restricted number of reuses
When creating a UDI record for reusable devices, you must include information about the number of reuses.
Where the device has a maximum number of reuses, you must provide this information in the field ‘Restricted number of reuses.’
Where the number of reuses is not restricted to a maximum number, you select ‘N/A’ in the field ‘For single use?’ This indicates that while the device is reusable, the number of reuses is not restricted.
Reusable devices where ‘N/A’ would apply includes:
- capital equipment
- devices that are restricted based on lifetime or shelf life of a device rather than a set number of reuses
- devices that are consumed during use, and the smallest trade item comprises a maximum number of tests greater than one
- devices supplied via Surgical Loan Kits (SLKs), where the manufacturer does not enforce any restrictions on the number of reuses and provides instructions on how to detect signs of material degradation.
Capital equipment
Capital equipment must meet UDI requirements.
As industry informally refers to and defines capital equipment as large, durable medical devices that are repeatedly used over time, for example MRI machines or CT scanners, the UDI requirements for these types of devices are grouped here.
This grouping reflects operational practice rather than regulatory terminology. For regulatory purposes, capital equipment is assessed under the general definition of a medical device in the Therapeutic Goods Act 1989.
You must label capital equipment devices with a UDI on the equipment itself, however, is not required to be directly marked. The UDI Carrier may be applied as a sticker, plate, printed directly onto the equipment itself, or be in any other form that can withstand the normal use and cleaning of the equipment.
Catherine the manufacturer
Catherine manufactures MRI machines. As capital equipment, they are in scope of UDI requirements and must bear a UDI.Catherine applies a metal plate with the UDI to the MRI machines, making them UDI compliant. As the labelling of her devices can withstand the normal use and cleaning, they do not require direct marking.
She may choose another form of label, such as a durable sticker, provided the UDI Carrier withstands normal cleaning processes for the device’s lifetime.
If the UDI-DI for the device changes, the label must be replaced with a new UDI-DI. If the UDI is presented through an electronic display, it is acceptable to display the new UDI-DI on an easily accessible screen such as an ‘About’ box.
Single use devices
Single use devices must meet UDI requirements.
A single use device is used on a single patient during a single procedure and then discarded. It is not intended to be reprocessed for use on another patient.
Generally, a single use device must bear a UDI on its label. However, where individual single use devices of a single model or version are distributed together in a single package, and intended to be stored in that package until removed for use:
- the UDI is not required on the device label
- the UDI must be applied to the device package.
This exception is not applicable to:
- any implantable device
- single use devices intended for individual commercial distribution
- where more than one model or version are distributed together in a single package.
When the end user (for example, healthcare provider) is not expected to have access to the package, the UDI Carrier should be on the individual device packaging. All other levels of packaging must bear a Package DI, except logistics units.
When the UDI is applied to a package containing multiple devices of the same model/version, the manufacturer must allocate a Unit of Use DI, unless all devices are intended for use by the same patient or user. In that case, Unit of Use is optional because it is assumed the patient or user will consume all the devices.
For more information on Unit of Use, see: Complying with the Unique Device Identification requirements for medical devices
Josh the manufacturer
Josh manufactures surgical gloves. Josh supplies his surgical in a box of 100.Josh’s surgical gloves are single use; however, they are intended to stay in the box until use. Josh is not required to apply a UDI to each individual glove in the box. To support traceability of the gloves, Josh is required to apply the UDI on the box.
In this scenario, Josh must also allocate a Unit of Use DI to his gloves.
Nick the manufacturer
Nick manufactures wound dressings for healthcare clinics. Nick supplies his wound dressings in a box of 10.Nick’s wound dressings are single use; however, they are expected to be removed from the base package before use. Because they are expected to be separated from the package bearing the UDI, Nick applies the UDI Carrier to each individual device.
Because the devices are individually labelled with the UDI, he does not need to allocate a Unit of Use DI. If Nick is unable to apply the UDI Carrier to each individual wound dressing, he must allocate a Unit of Use DI.
Personalised Medical Devices (PMDs)
Certain types of Personalised Medical Devices (PMDs) must meet UDI requirements.
PMDs are devices designed and manufactured, or adapted or modified, to meet the needs of an individual.
We use 3 specific terms to describe personalised medical devices:
- patient-matched medical devices
- adaptable medical devices
- custom-made medical devices.
For guidance specific to PMDs, see Personalised medical devices.
You must meet UDI requirements for:
- adaptable medical devices
- patient-matched medical devices.
You do not need to meet UDI requirements for custom-made medical devices.
Patient-matched medical devices (PMMDs)
PMMDs are exempt from inclusion in the ARTG until 1 July 2029. As UDI requirements only apply to devices included in the ARTG, PMMDs are exempt from UDI requirements until they are included.
Once your PMMD is included in the ARTG, you must meet UDI requirements for your PMMD - if it is in scope of the UDI requirements.
If your PMMD is eligible for the low volume exemption or specified articles exemption, your PMMD is exempt from UDI requirements.
Adaptable medical devices
Adaptable medical devices are personalised after they are manufactured.
They are modified, adapted or assembled to suit a specific individual. This is done in line with the manufacturer’s instructions.
Examples include:
- a limb prosthesis assembled from pre-manufactured components in line with the manufacturer’s instructions
- an off-the-shelf bruxism mouthguard, to be moulded by the patient themselves
- a prefabricated (non-prescription) orthotic insole, shaped and trimmed to the right size for a patient.
Adaptable medical devices need to be included in the ARTG and must meet UDI requirements, if they are in scope of devices required to meet UDI requirements.
Custom-made medical devices
A custom-made medical device (CMMD) is designed and made for a particular person. Unlike a PMMD, a CMMD is rare and unique, and the manufacturer cannot adequately and fully validate the design or production processes used. A health professional may determine that a CMMD is needed when no other device like it is included in the ARTG.
An example is an acetabular implant made for a person who is 2.26 metres tall and weighs 160 kilograms. The dimensions and tolerances of the implant needed to suit this person are outside the manufacturer’s usual design envelope. The implant is genuinely a ‘once-off’ and therefore is a CMMD.
Custom-made medical devices are exempt from UDI requirements.
Dental devices
Dental devices must meet UDI requirements.
Dental practitioners
As a registered dental practitioner, you have regulatory responsibilities to the TGA when:
- importing dental devices (a sponsor)
- manufacturing dental devices away from chair-side (a manufacturer and a sponsor).
For more information about regulatory responsibilities for dental practitioners, including information on what makes a dental practitioner a sponsor see Understanding requirements for dental practitioners when making and adapting personalised medical devices.
If these cases apply to you, you must meet UDI requirements for dental devices where:
- you are the sponsor of the dental device, and
- the dental device is in scope of devices that must meet UDI requirements.
You do not need to meet UDI requirements if you buy:
- finished devices from an Australian-based sponsor
- materials and components to manufacture non-implantable dental medical devices from an Australian-based sponsor who has already met UDI requirements.
Implantable dental devices
As a sponsor, or a dental practitioner acting as a sponsor, you must meet UDI requirements for implantable dental devices.
Implantable dental devices are not exempt from ARTG inclusion or UDI requirements when made using materials included in the ARTG. Examples of implantable dental devices include:
- dental implants and implant abutments
- implant abutments with special attachments
- temporary anchorage devices (TADS), such as Mini screws.
Dental devices attached to implant abutments or fixed to TADS are exempt from ARTG inclusion and UDI requirements when made using ARTG included materials. Examples include:
- crowns
- bridges
- fixed or removable non-implant orthodontic appliances.
Medical device and IVD device software
Medical device and IVD software must meet UDI requirements.
This includes:
- Software as a Medical Device (SaMD)
- medical devices incorporating software.
Software incorporated or embedded into a device does not need its own UDI if:
- it is not supplied as a standalone device
- it is not commercially available on its own.
The table below summarises software types that must meet UDI requirements.
| Type | Requires UDI? | AusUDID submission |
|---|---|---|
| Software as a Medical Device | Yes, if in scope of devices required to meet UDI requirements | Submit in UDI record as ‘Software as a medical device’. |
| Medical device incorporating software | Yes, if in scope of devices required to meet UDI requirements | Submit in UDI record as ‘Medical device incorporating software’. |
| Software incorporated into a medical device | No, if not standalone | No individual UDI record required. |
Medical devices incorporating software
Allocating a UDI
The manufacturer must allocate a UDI to the physical medical device that incorporates software. The embedded or incorporated software does not require its own UDI unless it is also supplied as a standalone product.
Kobe the manufacturer
Kobe manufactures an infusion pump with embedded software. He allocates a UDI to the physical device. He does not allocate a separate UDI to the software because it is not supplied on its own.Kobe submits the UDI record for the infusion pump to the AusUDID and records ‘Medical device incorporating software’ in the data field titled ‘Is the medical device software or does it incorporate software?’.
Medical devices incorporating software UDI placement
The UDI Carrier must be applied to the physical device’s label and packaging according to standard UDI requirements. If the device includes a user interface, you should also display the UDI in an accessible location such as an ‘About’ screen.
Software as a Medical Device (SaMD)
Allocating a UDI
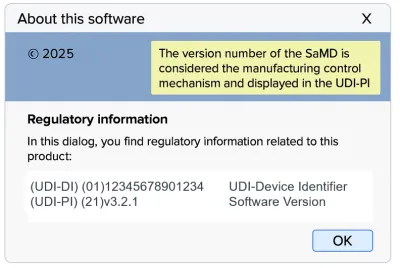
The manufacturer must allocate the UDI-DI at the system level of the SaMD. The version number acts as the manufacturing control mechanism and must be included in the UDI-PI.
Kevin the manufacturer
Kevin develops a diagnostic app classified as SaMD. Kevin allocates a UDI at the system level of the app. He includes the version number in the UDI-PI.Kevin’s UDI for his SaMD:
(01)12345678901234(21)v.3.2.1(17)251118.
Kevin displays the UDI in the app’s ‘About’ screen.
SaMD UDI placement
If you supply SaMD in a physical medium, for example a DVD, standard UDI labelling requirements apply:
- apply the UDI Carrier to the base package
- apply the UDI Carrier with unique Package DI(s) to all applicable higher levels of packaging.
The UDI-DI that is applied to the base package of the physical medium containing the SaMD and its packaging must be identical to the UDI-DI allocated to the system level SaMD.
If you supply SaMD in an electronic format, for example an app, you must display the UDI on screen a readily accessible by the user in an easily readable format. For example, an ‘About’ file or startup screen. When the UDI is provided via an electronic display, only the HRI form is required. You do not need to include the AIDC form in the electronic displays.
The image below shows how a UDI may be displayed in a pop-up box.
If your SaMD lacks a user interface, it must be capable of transmitting the UDI through an Application Programming Interface (API) or similar.
You include the Data Delimiter in the HRI form. This helps the end user to identify the UDI and determine which standard you have used to create the UDI.
If you label your SaMD with a physical UDI Carrier but later perform an upgrade that requires your device to be assigned a new UDI-DI, you may provide this new UDI-DI through the electronic display method. We recommend you inform the end user to remove the physical UDI Carrier to minimise confusion.
Software UDI triggers
SaMD version changes
You must allocate a new UDI-DI if a major software revision significantly changes:
- the original performance and effectiveness
- the safety or intended use of the SaMD.
These changes include but are not limited to:
- new or modified algorithms
- database structures
- operating platform
- architecture
- new user interfaces
- new channels for interoperability.
You must also submit a new UDI record when your SaMD is allocated a new UDI-DI.
You must allocate a new UDI-PI for minor revisions such as:
- bug fixes
- usability enhancements (not for safety purpose)
- security patches
- operating efficiency.
You should identify minor revisions by manufacturer specific identification methods such as:
- version
- revision number
- serial number.
We recommend that you record changes and how changes are communicated to users of the SaMD in your quality management system. Note that minor changes to software do not need to be recorded in the AusUDID.
Devices principally sold in retail
Devices principally sold in retail must meet UDI requirements.
Devices ‘principally sold in retail’ are those intended for sale in retail premises such as supermarkets and retail pharmacies. These devices are eligible for reduced labelling requirements.
The manufacturer must determine whether a device is principally sold in retail and must justify this if requested. The manufacturer may choose to liaise with their sponsor(s) to determine whether the device will be principally sold in retail.
If your device is supplied in both retail and healthcare settings, however, is predominantly supplied in healthcare settings, you must meet full UDI labelling requirements for this device. If your device is supplied in both retail premises and healthcare settings, you must meet full UDI requirements where the device is predominantly supplied in healthcare settings.
Example: An adhesive bandage is sold through a national Supermarket. This device is considered principally sold in retail. If the bandage is also used in medical centres as a secondary supply purpose, but the main supply destination is retail, this device still qualifies as principally sold in retail.
Reduced labelling requirements for devices principally sold in retail
Devices principally sold in retail has flexibility in the labelling of the trade level of packaging for the device:
- UDI-DI
- must be in human-readable form, however, does not need to be HRI
- must be in a machine-readable form, however you may use standards that better support retail use rather than the more comprehensive healthcare standards.
- UDI-PI
- must be in human-readable form, however, does not need to be HRI
- not required in machine-readable form, but you may choose to do so.
- production identifiers provided on the label may vary depending on the medical device type and current practice, but at least one production identifier must be included on the label.
- Unit of Use DI
- Because of their nature, devices principally sold in retail premises are exempt from Unit of Use requirements.
- Device Count for devices principally sold in retail should be set to ‘1’ in the AusUDID, as these devices are intended for a single user.
- Direct Marking DI
- Because of their nature, devices principally sold in retail premises are exempt from direct marking requirements.
The image below shows an example of a UDI compliant label for devices principally sold in retail.

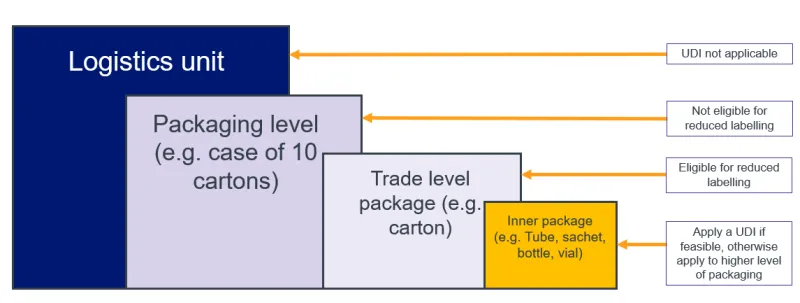
The image below shows how UDI applies to packaging levels for devices principally sold in retail. In this example:
- UDI is not applicable to logistics units, as they are not required to meet UDI requirements
- the packaging level must bear full UDI labelling
- the trade level is eligible for reduced labelling
- if multiple devices are contained in the trade level, apply the UDI to the individual devices if possible. If not feasible, apply to the higher level of packaging – in this case, the trade level.
Note that Unit of Use and direct marking are not applicable to retail devices.
Example of retail packaging levels.
The table below summarises the UDI-DI and UDI-PI requirements for devices principally sold in retail.
| Carrier form | UDI-DI | UDI-PI |
|---|---|---|
| HRI | Optional | Optional |
| Non-HRI | Required* | Required* |
| Machine readable or AIDC | Required | Optional |
You do not need to include both HRI and non-HRI if you choose to use HRI form.
You must meet all UDI requirements for all other applicable levels of packaging. Higher levels of packaging are not eligible for the reduced labelling.
Retail devices redirected to healthcare settings
There are some scenarios where devices principally sold in retail are redirected to healthcare settings.
Devices temporarily redirected due to shortages
In some situations, retail devices may be temporarily redirected to hospitals or healthcare providers. This can happen due to stock shortages, hospital purchasing decisions or similar circumstances. In these cases, it is not necessary to relabel the devices with the full UDI compliant labelling as this may create unnecessary burden and impact the supply of the products. These devices remain eligible for the reduced labelling requirements.
Devices redirected due to business changes
If a device that was originally intended for retail sale is supplied to healthcare settings on a more permanent basis, then full UDI labelling will be required on the device. This will ensure the device supports the traceability that is required for clinical environments.
Devices that frequently change supply channels
For devices that frequently shift between retail and healthcare supply channels, it is recommended that manufacturers apply full UDI labelling for these devices. This approach helps ensure consistent compliance and reduces the need for relabelling as distribution patterns change.
Aiden the manufacturer and sponsor
Aidan manufactures and supplies wound dressings to retail premises and medical centres. Most go to retail premises, so Aidan applies reduced UDI labelling:
- UDI-DI in non-HRI
- UDI-DI in machine readable
- UDI-PI in non-HRI.
If Aidan begins to supply the device predominantly in hospitals or healthcare settings, Aidan will need to change his labelling to be fully UDI compliant prior to supply in these settings.
Aidan may choose to use fully UDI compliant labelling for devices he sells in retail, so that any changes to where he supplies his device will not impact his ability to supply the device.
Refurbished devices, reprocessed single use devices, own brand or private labellers
UDI requirements for refurbished, reprocessed and relabelled devices depends on a range of factors, including:
- whether you are the original manufacturer of the device you are refurbishing or relabelling
- whether the device has undergone changes that have altered the intended purpose of the device
- whether the device that is being reprocessed is single use or reusable.
Note that reprocessing reusable devices in ways described in the manufacturer’s Instructions for Use does not make you the manufacturer. Reprocessing of reusable medical devices is part of the intended purpose for the medical device, and part of clinical practice. We do not regulate clinical practice.
Refurbished devices
Refurbishment is defined as a substantial rebuild from one or more used medical devices that may render it a ‘new’ medical device under the Regulations.
If you refurbish an already used medical device, you are considered the manufacturer of the device.
Your responsibilities as a manufacturer of a refurbished device depend on whether you are also the original manufacturer of the device.
Refurbishing a device originally manufactured by different manufacturer
If you refurbish a device that was originally manufactured by different manufacturer, you are considered the manufacturer of the refurbished device. In this scenario, you are required to allocate a UDI-DI to the refurbished device. You must meet all relevant UDI requirements for this device. Your device also requires an ARTG inclusion.
Refurbishers must create their own, new UDI for the refurbished medical device which will replace the original manufacturer’s UDI where it exists. This includes removing any UDI that has been directly marked onto the device.
Refurbishing your own device
If you refurbish a device that you originally manufactured, you may be able to resupply this device under the existing ARTG with the same UDI-DI, where:
- the refurbishment has not changed its intended purpose
- the device is considered the same model of device.
If you have changed the intended purpose of the device when refurbishing the device, you must allocate a new UDI-DI to this device. If the device is no longer considered to be within the set limits of specifications, performance, size and composition of the original model of device, it is considered a new model of device. In this case, you must allocate a new UDI-DI.
If you refurbish a device that you originally manufactured that is now required to meet UDI requirements, however this device does not yet have a UDI-DI allocated to it, you must allocate a UDI-DI to this device and meet all UDI requirements applicable. For example, if the device was previously not required to meet requirements as it was not mandatory at the time of manufacture, however the device is now in scope of UDI requirements at the time of remanufacture, you must meet UDI requirements for this device.
Angus the refurbisher
Angus refurbished a Class Is medical device that was originally manufactured by a different manufacturer to supply for reuse. In the process, Angus:
- stripped the device
- checked the components and replaced components not suitable for re-use
- assembled the device and tested the device against the original specifications of the device
- identified the device as a refurbished device.
Angus has certified the device is suitable for reuse. He has also assumed the legal liability for the quality, safety and performance of the device. Since he did these activities, Angus has become the manufacturer of the device and must now meet UDI requirements. This includes Angus allocating and applying the UDI to the device.
Angus is responsible for ensuring that the previous UDI on the device, if any, is removed entirely.
As Angus is also the sponsor of the device, he must meet the obligations of a sponsor. These include submitting the UDI-DI and related data to the AusUDID and linking the relevant ARTG to the UDI record.
Reprocessed single use devices
Reprocessing of single use devices makes them a new medical device and requires an ARTG and a UDI-DI. This includes refurbished single use devices and single use devices where the intended purpose has been altered, allowing the device to be reusable.
Note that some single use devices are marketed as non-sterile which require processing to make them sterile and ready for use. You are not considered the manufacturer if you process a single use device to prepare it for use, per the Instructions for Use.
Reprocessing a single use device originally manufactured by a different manufacturer
If you reprocess a single use device originally manufactured by a different manufacturer, you are considered the manufacturer of the single use device.
Reprocessing your own single use device
If you reprocess a single use device that you originally manufactured, and the intended purpose remains the same, you may be able to supply the reprocessed single use device under the existing ARTG entry and retain the existing UDI-DI. However, if the single use device intended purpose has changed, for example the device is now reusable, you must allocate a new UDI-DI as this is a UDI Trigger.
Own brand or private labellers
Own brand or private labellers who re-label a medical device are considered the manufacturer of these devices and are required to meet UDI requirements for these devices.
Relabelling devices under the manufacturer's instructions (sponsors and subcontractors) doesn't make you the manufacturer.
In vitro diagnostic (IVD) medical devices
IVD devices
IVDs must meet UDI requirements.
In most scenarios, IVDs share the same UDI requirements with medical devices.
IVD kits
Although the term ‘kit’ has a specific meaning in the Australian legislation, some products that are regulated as medical devices include the word kit in their name and include at least one medical device. For example, SARS-CoV antigen test kits, which only contain medical devices (including IVD devices).
You must meet UDI requirements for an IVD kit as well as for any components of the kit, where the component is:
- is sold separately from the kit
- has its own ARTG inclusion, and
- is in scope of devices required to meet UDI requirements.
If the kit does not include any components that are in scope of UDI requirements, you only need to meet UDI requirements for the kit itself.
If your IVD kit contains single use medical devices, these devices do not need a UDI where:
- the person(s) intending to use the device generally know the uses*
- the single use medical devices are not intended for individual use outside the context of the IVD kit, or
- they are not in scope of devices required to meet UDI requirements.
*You may be required to justify this on our request.
Because of their nature, the Unit of Use DI is not appropriate for IVD kits.
Placement of UDI on IVD kits
You should apply the UDI Carrier to the outside of the packaging. It must be readable or scannable whether placed on the outside of the IVD kit package or inside a transparent package.
Submission of IVD kit data to the AusUDID
As a sponsor, you are responsible for ensuring that the following data is supplied to the AusUDID:
- UDI-DI and related data of the IVD kit
- UDI-DI of each component of the IVD kit that has a UDI.
You are not required to link the UDI record for the components to the UDI record of the IVD kit.
Brooke the refurbisher
Brooke manufactures a Rapid Antigen Test Kit. Brooke’s RAT kit is a Class 3 IVD and is in scope of devices that must meet UDI requirements.Brooke does not supply any of the components of the IVD kit separately and all components are single use.
Brooke must allocate and apply a UDI to the IVD kit itself, but Brooke is not required to allocate and apply a UDI to any of the individual components.
Implantable devices
Because of the high risks with implantable devices, the UDI for an implantable device should be identifiable and able to be recorded before implantation. This can minimise the risks of misidentification of the implanted device.
The following UDI requirements apply for implantable devices:
- all base packages of implantable devices need to be identified by checking the UDI before surgery and captured at the point of implantation
- the UDI must be in both HRI and AIDC formats
- the UDI-PI should include:
- for an active implantable device, the serial number
- for other implantable devices, the serial number or lot number per the manufacturer's quality management system.
You do not need to directly mark the UDI on implantable devices.
Examples of labels that you may use to support identifying implantable devices include:
- a tear-away tag bearing the UDI
- peel-off labels bearing the UDI affixed to autoclave box holding the implantable device.
Surgical Loan Kits (SLK)
SLKs are exempt from UDI requirements at the kit level; however, component medical devices in the SLK must meet UDI requirements.
SLKs typically include reusable surgical instruments, implantable medical devices, or single-use medical devices that are supplied on loan or long-term consignment to hospitals for specific surgical procedures. They can be customised for the hospital, procedure, surgeon, or patient.
SLKs are assembled by an SLK manufacturer using medical devices, also known as component medical devices, that were manufactured by component manufacturers and included in the Australian Register of Therapeutic Goods (ARTG) by component sponsors.
Component manufacturer
The component manufacturer is the entity that manufactures the individual component medical devices included in a SLK. They are responsible for design, production, packaging, labelling, and assigning the intended purpose to the component medical device. The component manufacturer is responsible for ensuring each component medical device is allocated a UDI-DI and UDI-PI and meets any other UDI labelling requirements applicable.
Component sponsor
The component sponsor is the entity that includes the component medical device in the ARTG for supply in the Australian market. The component sponsor is responsible for ensuring a UDI record for each component medical device they supply is submitted to the AusUDID.
SLK manufacturer
The SLK manufacturer is the entity that assembles, packages, or labels the kit using ready-made component medical devices from component manufacturers and arranges distribution to hospitals.
The SLK manufacturer is responsible for providing UDIs to the hospital when supplying the SLK.
An entity may hold several roles. For example, an entity may be both the component sponsor and the SLK manufacturer.
SLK at kit-level
SLKs are exempt from UDI requirements at the kit level. The tray, tub, case, or caddy used for transport is considered a logistics unit which is not required to meet UDI requirements.
SLK components
Component medical devices in an SLK are not exempt from UDI requirements. All component medical devices must meet UDI requirements if in scope of UDI requirements.
As a component manufacturer, you must assign a UDI including the UDI-DI and UDI-PI to each model of device.
As a sponsor, you must ensure that a UDI record is submitted to the AusUDID for each model of device.
Providing UDI for SLK components
As a component manufacturer, you must provide the full UDI for each device to the SLK manufacturer.
With some exceptions for small non-sterile devices, the SLK manufacturer must provide the full UDI for each component medical device in the kit to the hospital when they supply the SLK, ready to be easily accessible at the point of care.
We do not prescribe a specific method for providing the UDI for SLK components. Component manufacturers and SLK manufacturers may adopt flexible approaches consistent with international regulators to provide the UDIs to the SLK manufacturer and hospital. These methods include:
- stickers
- tags
- inventory sheets
- data carrier strips.
Ruby the component manufacturer
Ruby manufacturers component medical devices. She must meet UDI requirements for all medical devices that are Class Is, Class IIa, Class IIb, or Class III.Ruby provides the full UDI for each applicable device to Emily.
Emily the SLK manufacturer
Emily assembles SLKs. Each SLK includes reusable surgical instruments, implantable medical devices and single-use devices. Emily ensures each component medical device is included in the ARTG and meets UDI requirements, where applicable.
Emily’s SLK contains 4 Class I medical devices and 10 Class III medical devices.
Emily must ensure each of the 10 Class III medical devices meet UDI requirements. However, UDI requirements do not apply to Class I medical devices.
Emily does not need a UDI for the SLK itself.
Emily provides the full UDI for each applicable to the hospital when she supplies the kit, with some exceptions for small non-sterile devices.
John the component sponsor
John is the sponsor of Ruby’s component medical devices. He must ensure that any medical devices that are Class Is, Class IIa, Class IIb, or Class III meet UDI requirements.
John must also ensure that a UDI record for each model of device is submitted to the AusUDID.
Small non-sterile devices
We recognise the challenges in providing and retaining production information with small non-sterile devices in SLKs. These challenges include:
- Loss of UDI Carrier during sterilisation
- Before surgery, small non-sterile devices are often sterilised and removed from their packaging or labelling, which could contain the UDI. Once separated from the UDI Carrier, the device may be difficult to identify later in the hospital or after the SLK is returned.
- Replenishment issues
- When small non-sterile devices are replenished in an SLK, they may come from different production batches. This makes it difficult to maintain accurate production information and track individual components.
- Direct marking impracticalities
- While direct marking is not required for implantable medical devices, industry advised that even if required, direct marking would be technically infeasible for small non-sterile devices due to their size.
To address these challenges, small non-sterile devices have reduced UDI requirements:
- SLK manufacturers must provide the UDI-DI for small non-sterile devices that are supplied in an SLKs
- SLK manufacturers are not required to provide the UDI-PI for small non-sterile devices that are supplied in an SLK.
All devices must still be allocated a UDI-DI, and the UDI-DI must be easily accessible at the point of care.
As a component manufacturer, you may voluntarily directly mark small non-sterile devices where technically feasible, enabling UDI-Pi availability at point of care.
The manufacturer is responsible for determining what is considered a small device. You may be required to justify this decision to the TGA, when requested.
Small non-sterile devices supplied in SLKs and outside SLKs
When a small non-sterile device is supplied both inside an SLK and outside an SLK, it must carry both the UDI-DI and the UDI-PI when supplied outside the SLK. The UDI Carrier for devices supplied outside an SLK should follow standard UDI labelling requirements. This does not change how the UDI is provided for devices within an SLK, where alternate methods are acceptable.
Ruby the component manufacturer
Ruby supplies small non-sterile devices both in an SLK and outside of an SLK.
For the devices supplied in an SLK, Ruby provides the full UDI to Emily the SLK manufacturer. When Emily assembles the SLK, she provides the hospital with the UDI-DI for the small non-sterile devices but not the UDI-PI.
For the devices supplied outside of an SLK, Ruby applies a UDI Carrier that includes both the UDI-DI and the UDI-PI per standard UDI labelling requirements.
Implications for market actions
Where the UDI-PI is not provided for small non-sterile devices in an SLK:
- any market action, device incident report, or adverse event notification cannot be limited to a subset of production
- actions must include all production of the device with the relevant UDI-DI
- SLK manufacturers must extend regulatory actions to all SLKs containing devices with that UDI-DI.
Where the UDI-PI is not provided for small non-sterile devices in an SLK:
- any market action, device incident report, or adverse event notification cannot be limited to a subset of production
- actions must include all production of the device with the relevant UDI-DI
- SLK manufacturers must extend regulatory actions to all SLKs containing devices with that UDI-DI.
Arabella the SLK manufacturer
Arabella recalls a screw type that is supplied in multiple SLKs. Because the UDI-PI was not provided to hospitals, Arabella must recall all screws with that UDI-DI. She cannot recall individual batches.
The table below provides a summary of UDI requirements for SLKs and components medical devices.
| Item | UDI required? | Details |
|---|---|---|
| SLK kit (tray, tub, case, caddy) | No |
|
| Component devices | Yes, if in scope of UDI requirements |
|
| Small non-sterile devices supplied in an SLK | Partial |
|
| Method of UDI provision | Flexible |
|
System or Procedure Packs (SOPPs)
System or Procedure Packs (SOPPs) must meet UDI requirements.
SOPPs at pack-level
You must allocate a UDI to a SOPP if it contains one or more medical device in scope of UDI requirements.
Your SOPP does not need a UDI on the pack if it only contains:
- Class I non-sterile non-measuring medical devices, or
- Class I measuring (Im) medical devices.
However, the assembler or manufacturer of the SOPP may choose to apply a UDI voluntarily.
SOPP components
Any component of a SOPP that is a medical device that is in scope of UDI requirements must have a UDI assigned for the component unless it is:
- not commercially available on its own
- an individual single-use disposable device where:
- the person(s) using them generally know the uses, and
- the device is not intended for individual use outside the context of the SOPP. For example, an unpackaged sterile syringe in a sterile pack cannot be used for another procedure once removed from the pack
- exempt from UDI requirements.
Medical device components that you supply separately from the SOPP must:
- be included in the ARTG
- comply with UDI requirements, unless exempt.
Breanna the manufacturer
Breanna manufactures a SOPP that includes:
- 3 Class I medical devices
- 3 Class Is medical devices
- 3 Class IIb medical devices.
Because the pack contains devices in scope of UDI requirements, Breanna allocates a UDI to the SOPP.
For the individual components in the SOPP:
- Breanna does not need to apply a UDI to the 3 Class I medical devices, as Class I medical devices are not in scope of UDI requirements.
- Of the 3 Class Is medical devices, 2 are individual single-use devices. Breanna does not supply these separately, and they are discarded once the SOPP is opened. These devices do not need their own UDI.
- The third Class Is medical device is supplied both inside the SOPP and separately. Breanna must allocate a UDI to this device.
Of the 3 Class IIb medical devices, Breanna supplies one separately from the SOPP. Only this Class IIb device needs its own UDI.
Systems that are medical devices
Systems that are medical devices must meet UDI requirements.
A system is 2 or more goods that the manufacturer intends to be connected, used together or combined to achieve a specific medical purpose. The goods may be packaged together or separately.
A system is not:
- a single item
- miscellaneous items that the manufacturer does not intend to be used together for a specific medical purpose
- bulk packs of one or more items
- a procedure pack (though a procedure pack can include a system in it).
Examples of systems that are medical devices include:
- knee-joint replacement system
- orthopaedic drill system
- a patient-monitoring system with a monitor, power cable, and backup power supply
- a blood-glucose monitoring kit with a blood-glucose meter, test strips, controls, lancets, and a lancing device.
As a manufacturer, you must allocate a UDI to the system that is a medical device.
As a sponsor, you must submit a UDI record for the system to the AusUDID.
Where all individual medical devices in the system comply with UDI requirements on their own, you are not required to allocate a UDI to the system as a whole; however, you may choose to do so. UDI requirements for systems that are medical devices do not supersede or override any existing requirements, including Instructions for Use requirements.
Systems that are configurable
System UDI
For systems that can be configured, you must allocate a UDI to the entire system. This UDI is referred to as the System UDI. It is not an additional identifier; it simply a term used to distinguish and reference the complete system.
As a manufacturer, you should place the UDI Carrier for the System on the assembly that most likely does not get exchanged in its lifetime.
The System UDI will comprise both a UDI-DI and UDI-PI.
System UDI-DI
As a manufacturer, you must allocate a UDI-DI to defined groups of configurations the same way you allocate a UDI-DI to defined models of medical devices. This UDI-DI is referred to as the System UDI-DI. Again, this is not an additional identifier, but a distinction used to identify configuration groups.
As a manufacturer, you are responsible for defining the groups as the collection of possible configurations for a given product line as described in a regulatory file.
System UDI-PI
You must allocate a System UDI-PI to each individual system. A later change of a component, sub-system or accessory of the system does not change the System UDI-PI.
The concept of System UDI, System UDI-DI and System UDI-PI is to help define configurations of systems. You are not required to provide an additional identifier, nor are you required to enter an additional identifier into the AusUDID. The System UDI-DI is the Primary DI for configurable systems.
Example of an upgrade to a system which impacts safety or performance
Claire the manufacturer – MRI system
Claire manufactures an MRI system, 'Model A', and distributes it to customers.
She develops new features and functionality for that MRI system which her original approved specifications do not cover. This could be:
- hardware
- software
- a combination of both.
These changes alter the safety profile, performance or intended use of the system.
Claire determines this results in a new model, Model B.
If Claire decides to modify the device as a new installation, she must give the modified device a new System UDI-DI. Alternatively, she may supply an upgrade kit as a separate medical device with its own UDI-DI. The UDI-DI of the upgrade kit and the original System UDI-DI together identify the changed device.
Example of a component change which impacts safety or performance
Alice the manufacturer – X-ray system
Alice manufactures an X-ray system with a 50 kV generator. She replaces the 50kV generator with a 100 kV generator, which is not specified in the original configuration and changes system performance.
Alice determines this creates a new model or version. If she modifies the device as a new installation, she must allocate a new System UDI-DI. Alternatively, she may provide an upgrade kit as a separate medical device with its own UDI-DI. The UDI-DI of the upgrade kit and the original System UDI-DI together identify the changed device.
Example of a new diagnostic feature, not previously approved, added to device
Bella the manufacturer – Cardiac ultrasound system
Bella manufacturers a cardiac ultrasound system. She introduces a diagnostic algorithm that enables new data calculations and imaging options. The algorithm introduces new indications for use and changes system features and performance.
Bella determines this creates a new model or version. If she modifies the device as a new installation, she must allocate a new System UDI-DI. Alternatively, she may provide an upgrade kit as a separate medical device with its own UDI-DI. The UDI-DI of the upgrade kit and the original System UDI-DI together identify the changed device.
As a manufacturer, you do not need to allocate a new UDI-DI when changes do not require the device to be distinguished from the original.
Example of a system component changed in an installed device, no change in safety or performance
Indigo the manufacturer – CT system
Indigo manufactures a CT System. One of Indigo’s installed CT Systems has an X-ray tube which has reached the end of its life. Indigo replaces this tube with a newer model tube without other changes to the device or its labelling.
Safety, performance and intended use remain unchanged. As such, Indigo determines this is not a new model, and the System UDI-DI does not change.
Example of a customer-selectable option changed for an installed device
Olivia the manufacturer – CT system
Olivia manufactures a CT System with an ARTG inclusion that covers several diagnostic algorithms. Customers choose which algorithms they would like to activate when ordering. A customer later activates an approved algorithm on an installed system because of their changing business needs.
The extra algorithm may be installed or activated and does not change the model or version of the system. As such, the System UDI-DI does not change.
Example of an addition of an accessory for an installed device
Matilda the manufacturer – System used with accessories
Matilda manufactures a system designed for use with accessories.
Customers adding or using accessories with the System is covered by the original specifications for the defined groups of configurations and does not change the model or version of the system. As such, the System UDI-DI does not change.
System components or accessories
As a manufacturer you must allocate a UDI to each component, sub-system, consumable or accessory if it:
- is a medical device in its own right
- is commercially available on its own
- is included on the ARTG, and
- is in scope of devices required to meet UDI requirements.
For example, an Automated External Defibrillator (AED) supplied with electrodes and batteries must be included in the AusUDID as a system. If the batteries and electrodes require their own UDI, you must include them in the AusUDID as separate devices.
We recommend you link the UDI record(s) of the components or accessories to the ARTG ID for the System you supply them against, as well as their own ARTG ID.
Maddie the sponsor – System used with accessories that are sold separately
Maddie supplies a system that customers can use with accessories. Maddie also supplies the accessories separately. The accessories have a different ARTG inclusion from the system. In this case, Maddie:
- submits a UDI record for the system and links it to the ARTG inclusion for the system
- submits a UDI record for each applicable accessory, and links it to the ARTG inclusion for each accessory and the ARTG inclusion for the system.
IVD systems
Many IVD systems use test reagents and accessories that are higher-class medical devices than the instrument they are used with. You must include the reagents and accessories in the AusUDID separately from the instrument.
Medical device accessories
Medical device accessories must meet UDI requirements in certain circumstances.
An accessory is a device intended to support, supplement or augment the performance of one or more parent devices. The accessory allows or helps the parent device to be used in a way that the manufacturer of the device intended.
Accessories must have their own UDI where the accessory:
- is individually labelled, packaged and sold separately from the parent device
- has its own ARTG inclusion (i.e. is not regulated under the parent device’s ARTG inclusion)
- is in scope of devices required to meet UDI requirements.
Accessories do not need to meet UDI requirements where the accessory:
- is packaged and sold with a parent device that has its own UDI
- is exempt from inclusion in the ARTG, or
- is otherwise exempt from UDI requirements.
Replacement parts or spare parts
Replacement parts or spare parts must meet UDI requirements in certain circumstances.
A replacement part or spare part is not a finished device in its own right. These are not considered an accessory for a device, as they would not normally be supplied separately for use with the device.
You are only required to meet UDI requirements for replacement parts or spare parts where the part:
- is sold separately from the device
- has its own ARTG inclusion, and
- is in scope of devices required to meet UDI requirements.
Amanda the sponsor and manufacturer
Amanda is both the manufacturer and sponsor of a system. A component in the system reaches the end of its life, and her customer orders a replacement. Amanda sells this component separately as well as part of the system. The component has its own ARTG inclusion and is classified as Class Is. Because the component is sold separately, has its own ARTG inclusion and falls within the scope of devices required to meet UDI requirements, Amanda allocates a UDI to the component.
The customer also requests a new screw for the system because the existing screw has lost its threading. Amanda does not sell screws separately, they do not have an ARTG inclusion, and they are not medical devices. Therefore, screws are not in scope for UDI requirements. Amanda provides the screw to the customer, but it does not require a UDI.
Contact lenses
Contact lenses must meet UDI requirements where they are included in the ARTG.
Many contact lenses are not currently included in the ARTG because of they are subject to the Patient Matched Medical Devices transition period. Devices eligible for this transition are exempt from UDI requirements until 1 July 2029.
You may choose to meet some UDI requirements before this date; however, you cannot meet UDI record submission requirements until your devices are included in the ARTG. UDI record submission requires linkage to an ARTG entry. Until then, you can only meet UDI labelling requirements.
UDI allocation for contact lenses
You must allocate a unique UDI-DI for each device model. Each production run with different production identifiers must have a separate UDI-PI.
You may allocate the UDI-DI to align with a defined design envelope and use the UDI-PI to track specific quality combinations.
Applying a broader definition of model will affect actions for specific devices. Any market action, such as a recall or correction, would need to apply to all devices covered by that broader definition.
The number of UDI-DIs depends on how you define and manage models. For example:
- you may allocate one UDI-DI per design and use the UDI-PI to track materials and colours
- You may allocate a separate UDI-DI for each design, material or colour combination.
As the manufacturer, you are responsible for determining the design parameters and envelope for your contact lenses. You must justify this decision if requested.
Boundary products
Boundary products must meet UDI requirements where the product is:
- Included in the ARTG as a medical device
- In scope of devices required to meet UDI requirements.
Examples include:
- Eye lubricating products that are only intended to lubricate the eye and do not have pharmacological components intended for antiseptic effect
- Liquid, spray, wipe or aerosol disinfectants to be used for disinfecting medical devices.
For guidance specific to boundary products, see: Understanding rules for boundary and combination products.
Combination products
Combination products must meet UDI requirements where the product is:
- Included in the ARTG as a medical device
- In scope of devices required to meet UDI requirements.
Examples include:
- Medical devices used to administer medicine that is supplied separately, such as syringes.
For guidance specific to combination products, see: Understanding rules for boundary and combination products.
Co-packaged medical devices
Co-packaged medical devices must meet UDI requirements where the product is:
- included in the ARTG as a medical device
- in scope of devices required to meet UDI requirements.
It is important to note that co-packaged medical devices are regulated differently depending on the regulatory jurisdiction. Where the device is regulated in Australia as a medical device, it must meet UDI requirements, regardless of the device containing a medicine component.
Where the device also bears medicine serialisation, we encourage manufacturers to include the UDI ISO symbol to clearly distinguish the UDI.
UDI ISO symbol
