Prescription medicines registration process
Guidance on the process and regulatory requirements to register prescription medicines.
Purpose
This guidance provides an overview of our registration process for applications that need to be supported by nonclinical, clinical and/or bioequivalence data (category 1 and category 2).
Applications to vary a product registration (section 9D of the Therapeutic Goods Act 1989 (the Act)) and not supported by nonclinical, clinical and/or bioequivalence data are outside the scope of this document.
Introduction
The TGA registration process for prescription medicine applications, that need to be supported by nonclinical, clinical and/or bioequivalence data (category 1 and category 2). This regulatory process is designed to improve the efficiency and timeliness of the registration of prescription medicines without compromising the scientific rigour of the evaluation process, thus ensuring the maintenance of appropriate standards of quality, safety, and efficacy. This document describes this process and outlines the relevant regulatory requirements.
The key elements of this process are:
- management by milestones
- an improved quality of dossiers prepared in accordance with common technical document (CTD) format and other TGA regulatory requirements
- a pre-submission planning phase where applicants lodge details of a proposed application at least 2¼ months prior to lodgement of the dossier allowing the TGA to identify milestone dates and plan resource requirements (this is not required for submissions lodged in eCTD format if the sponsor selects the PPF-only option)
- a submission phase where the applicant must lodge a complete dossier, there being no opportunity to deliver new data after the submission date except as required by the Therapeutic Goods Act 1989 (the Act)
- requests for information under section 31 of the Act are consolidated and issued at the end of the initial evaluation phase.
Legislative instruments made under section 9D and section 23 of the Act support this process. These instruments provide the legislative basis for the documents that specify the regulatory requirements for applications lodged under this process.
Overview
Scope
This document provides an overview of the TGA's registration process for applications that need to be supported by nonclinical, clinical and/or bioequivalence data (category 1 and category 2).
Applications to vary a product registration (section 9D of the Act) and not supported by nonclinical, clinical and/or bioequivalence data are outside the scope of this document.
Regulatory and supporting documents
Category 1 and 2 applications for new registrations are made under section 23 of the Act. Section 23 requires that applications are made in a form approved by the Secretary. The currently approved form is the CTD format.
Category 1 and 2 requests to vary the entry in the Australian Register of Therapeutic Goods (ARTG) of registered therapeutic goods are made under section 9D of the Act. Section 9D requires that applications are made in a manner approved by the Secretary. The currently approved manner is the CTD format.
The CTD format is described in the following documents:
- CTD Module 1: Administrative information and prescribing information for Australia
- ICH M4Q Common technical document for the registration of pharmaceuticals for human use - Quality (CPMP/ICH/2887/99 Rev 1 Quality)
- ICH M4S Common technical document for the registration of pharmaceuticals for human use - Safety (CPMP/ICH/2997/99 Rev 1 Safety)
- ICH M4E Common technical document for the registration of pharmaceuticals for human use - Efficacy (CPMP/ICH/2887/99 Rev 1 Efficacy).
For category 1 and 2 applications, other than applications solely for an additional trade name, the section 23 and section 9D instruments specify applications must comply with the following regulatory documents:
- Mandatory requirements for an effective application
- Pre-submission planning form
- prescription medicines (PREMIER) electronic lodgement facility (for applications to register a new chemical entity, new fixed combination, similar biological medicinal product or a new generic medicine) or
- the form Application for the registration, or to vary the conditions of registration, of prescription medicines (all other applications)
In addition to the documents specified by the section 9D and section 23 instruments, the TGA has produced other documents which provide further assistance for applicants who are lodging a PPF or dossier. These include:
- Prescription medicine registration process (this document)
- Information for applicants completing a pre-submission planning form
- Submit evaluation information to us electronically.
It is the applicant's responsibility to identify all application types that are included in a submission by selecting all applicable types in Part C of the application form.
Only those application types that are identified in the form will be evaluated for registration in the Australian Register of Therapeutic Goods (ARTG).
For example, if you are seeking an extension of indication and an additional change to the Product Information, a type C application under S23 and a separate type J application under 9D must be indicated in order for the applications to exist individually.
The highest fee category applies where multiple application types are included in a submission.
Key regulatory requirements
Management by milestones
The registration process consists of eight phases with eight milestones, allowing effective planning and tracking by the TGA and applicants. Each phase has established timeframes.
To facilitate management by milestones, all applications received in a given intake will proceed as a group through the phases and milestones, a process known as 'batch processing'.
Quality of applications
For the TGA to ensure that planned and target timeframes are met, it is critical that applicants lodge well-planned, high quality applications. Should a dossier not meet the regulatory requirements, it will be considered 'not effective' and will not be accepted for evaluation.
It is not possible for applicants who submit incomplete, delayed, or poor quality applications to provide unsolicited information during the regulatory process to rectify these deficiencies.
Pre-submission phase
Under the registration process, applicants provide the TGA with planning data in the Pre-submission planning form (PPF) at the pre-submission phase. Planning data include general submission information as well as information about the proposed application type and details of the quality, nonclinical and clinical evidence that will be provided in the dossier. The PPF provides the TGA with the necessary information for effective resource planning.
If the TGA considers a PPF complete and acceptable, the applicant will receive a Planning letter that provides key dates for the phases and milestones of the regulatory process for the application.
Comment
Pre-submission meetings may be requested where appropriate - the pre-submission phase does not replace the opportunity for applicants to conduct face-to-face discussions with the TGA regarding aspects of their proposed application. Pre-submission meetings may occur at any stage prior to PPF lodgement.
Meetings between TGA and applicants are conducted according to the processes described in the guideline Pre-submission meetings with the TGA. The TGA point of contact should be the PMAB Case Manager of the relevant clinical evaluation unit. Information on the areas of responsibility for the clinical units is included in Clinical evaluation sections for prescription medicines.
Submission phase
Applicants provide a dossier for evaluation at the submission phase. Applicants must ensure applications meet TGA regulatory requirements for format and content or the application will be considered 'not effective' and will not be accepted for evaluation. There will be no requests for information or documents under section 31 of the Act at the submission phase to address deficiencies in applications that do not meet TGA requirements.
Applications must be received by the date noted in the Planning letter to allow the TGA to complete the necessary administrative activities by the end of the same month.
At the end of the submission phase, the TGA will send the applicant a Notification letter advising that the application has either:
- been considered effective and is accepted for evaluation or
- been considered 'not effective' and is not accepted for evaluation.
Priority review designated applications
TGA now has a formal Priority review pathway for faster assessment of vital and life-saving prescription medicines for which a complete data dossier is available. The target timeframe of 150 working days is up to three months shorter than the standard prescription medicines registration process. A valid Priority review designation must be held in order to access the Priority review pathway. More information on Priority review is available at:
- Applying for priority determination for a prescription medicine
- Meeting the eligibility criteria for priority review determination
Provisional approval pathway
The TGA now has a formal Provisional approval pathway for the provisional registration of prescription medicines on the basis of preliminary clinical data, where there is the potential for a substantial benefit to Australian patients. A valid Provisional determination must be held in order to access this pathway.
More information on Provisional approval is available at:
- Provisional approval determination: A step-by-step guide for prescription medicines
- Provisional approval determination eligibility criteria: Including supporting documentation
- Provisional registration process: a step-by step guide for prescription medicines
- Provisional registration extension and transition to full registration
Information
Applications must contain all the information and material required for purposes of section 9D or section 23 of the Act and the documentation presented at submission will be taken as the complete application.
The applicant certifies in the PPF that the content of the complete dossier aligns with the information provided in the PPF and that the dossier, as received by the TGA, will be considered to be the full and complete dossier notwithstanding any further data requested by the TGA (including under section 31 of the Act) and/or new safety related data, which the applicant is obliged to bring to the TGA's attention. Applicants are strongly advised not to lodge a PPF before the full extent of the supporting data necessary for the application to be evaluated is known to be available.
To assist applicants prepare a complete application, TGA has published numerous documents and guidelines on the preparation and submission of prescription medicine applications. Applicants of prescription medicine applications should refer regularly to the TGA web site for those guidelines and policies relating to a particular application type of interest. Where a relevant guideline is not met, an appropriate justification must be provided in the dossier.
Once an applicant has submitted the dossier to TGA any data received by TGA without being requested by TGA will be considered 'unsolicited information'.
Data submitted after submission of the dossier must be accompanied by a covering letter which includes the submission number of the relevant application, the purpose of the correspondence and whether the information and material is 'solicited' or 'unsolicited'.
Solicited information
TGA considers the following to be solicited information:
- information and documents sent in response to the consolidated section 31 request following the first round assessment phase. The request will be sent on the date identified in the Notification letter allowing applicants time to conduct any necessary preparation activities.
- responses to informal requests from TGA staff for information or documents (for minor issues and clarifications). These requests may occur throughout the evaluation process and are intended only to maintain efficiency in the evaluation process, not to rectify deficiencies in incomplete or poor quality applications.
All solicited information in response to questions should be submitted in a question and answer format which is cross-referenced to replacement volumes where appropriate.
Applicants will also be able to:
- comment on the evaluation reports to highlight any errors of fact or major omissions
- comment (pre-ACM/ACV response) on the delegate’s request for advice from the independent Advisory Committee on Medicines (ACM) or Advisory Committee on Vaccines (ACV) when such advice is sought.
Unsolicited information
TGA will accept the following unsolicited information for evaluation:
- safety data (see 'safety data' section for the definition of safety data). Applicants are obligated to inform the TGA as soon as new safety data becomes available.
- new/renewed/updated TGA manufacturing licences (for Australian manufacturing sites) and good manufacturing practice (GMP) clearances (for overseas sites) issued for those sites identified in the application.
- revised trade names. An applicant may change, but not add, a trade name at any time prior to the end of the second round assessment phase.
In the event that the TGA receives other unsolicited information from an applicant, the applicant should be aware that there is a likelihood that the TGA will not evaluate the data. Applicants must, therefore, ensure that their dossier contains the full data set that they wish the TGA to evaluate and that the mandatory requirements (as applicable to the nature of the medicine and application type) are met.
The TGA will work with applicants of medicines of critical importance to the Australian community in emergency situations to ensure this policy does not adversely affect the timely evaluation of such medicines. Applicants of such medicines are encouraged to discuss arrangements with the TGA at a pre-submission meeting and in any case, as early as possible.
Safety data
For the purposes of the registration process, new safety data refers to information that might negatively influence the benefit-risk assessment of a medicinal product. Examples include but are not limited to:
- an increase in the rate of occurrence of an 'expected,' serious adverse drug reaction which is judged to be clinically important
- a significant hazard to the patient population, such as emergent lack of efficacy with a medicinal product used in treating a life-threatening disease, or a spate of unlisted reports of an uncommon or rare but serious adverse event
- a major safety finding from a newly completed animal study (such as an increased incidence of tumours in treated animals)
- pooled analyses of clinical studies that suggest significant dose or time dependent adverse effects that were neither apparent nor available at the time of lodging the application
- safety data associated with regulatory action in the event of, or to prevent risk to public health associated with use of the product in countries where it is already registered.
Safety data in this context do not include, for example:
- studies which only confirm information previously submitted to the TGA, including extension studies of submitted nonclinical or clinical studies (i.e. a, b and c above do not apply)
- studies or other data which support the use of the medicine in accordance with the proposed recommendations but which do not identify a new unexpected adverse drug event or an increased incidence of an expected adverse drug event
- pharmaceutic data
- nonclinical studies to justify proposed impurity limits.
Planning and tracking of applications
Regulatory phases
The regulatory process for evaluation of prescription medicines consists of eight phases. Each phase has a milestone that must be completed before commencement of the following phase. This approach allows effective and transparent management of resources and timelines for all applications.
| Phase | Relevant milestone | |
|---|---|---|
| 1 - Pre-submission | MS1 | Outcome of pre-submission planning sent |
| 2 - Submission | MS2 | Outcome of application consideration sent |
| 3 - First round assessment | MS3 | Outcome of first round assessment and section 31 request for information or documents sent |
| 4 - Consolidated section 31 request response | MS4 | End of section 31 request response period |
| 5 - Second round assessment | MS5 | Outcome of second round assessments sent |
| 6 - Expert advisory review | MS6 | Outcome of expert advisory committee review sent |
| 7 - Decision | MS7 | Decision made by delegate |
| 8 - Post-decision | MS8 | Administrative and regulatory activities complete |
Appendix 2 contains a pictorial representation of the regulatory phases. Section 5 of this document provides details about each of the phases.
Regulatory timelines
Depending on the type of application, different phases will be required which will affect the overall timeframe for the regulatory process. For initial planning purposes, applications are categorised into four distinct groups that are covered by some or all regulatory phases.
| Pre- submission | Submission | First round assessment | Consolidated section 31 response | Second round assessment | Expert advisory review | Decision | Post- decision | |
|---|---|---|---|---|---|---|---|---|
| New chemical entities, extensions of indications | Default plan | Default plan | Default plan | Default plan | Default plan | Default plan | Default plan | Default plan |
| Generic medicines | Default plan | Default plan | Default plan | Default plan | Default plan | Optional | Default plan | Default plan |
| PI changes, variations to existing products | Default plan | Default plan | Default plan | Default plan | Default plan | Optional | Default plan | Default plan |
| Additional trade names | Default plan | Default plan | Optional | Optional | Default plan | Default plan |
The registration process introduces planning timeframes into each of the phases, as indicated in appendix 4 and section 5 of this document. While the legislated TGA commitment of 40 working days between receipt of an application and notification of acceptance or rejection of that application remains, under the registration process this phase (the submission phase) will typically be completed in 15 or 21 calendar days, unless otherwise advised in the Planning letter.
The legislated TGA commitment of 255 working days between acceptance for evaluation through to the delegate's decision will also remain but will not be used for planning or for target times. The registration process is designed to take, on average, 330 calendar days (11 months), including the time for applicant activities.
Examples of timelines showing target milestone timeframes is provided in Appendix 1.
Submission planning
Planning for the evaluation process occurs during the pre-submission phase where the application type, the product, the scope and scale of the application, and the date of proposed dossier lodgement are identified. The information provided in the PPF allows the TGA to identify the resources required, including the specific areas of expertise, to evaluate an application. This phase also allows for the identification of any circumstances that may require the standard timeframe to be altered. Applicants will be notified of any alterations to the target timeframes.
Under the registration process, the TGA begins allocating resources for the evaluation phase when the PPF is lodged. These resources are based on the expected date of dossier lodgement. If a dossier does not arrive by the expected date, the TGA will reallocate the resources for that application and the applicant must re-commence the regulatory process from the beginning by lodging another PPF.
Submission tracking
Once the PPF is lodged, the TGA will track the application through the regulatory process. Each phase of the regulatory process concludes with the completion of a milestone.
At key points in the registration process TGA will notify the applicant of the evaluation plan milestone dates: an evaluation plan is included in the Planning letter at Milestone 1, the Notification letter at Milestone 2 and the Milestone 3 letter at Milestone 3. As target timeframes may change, applicants should always consult the most recent letter.
To facilitate tracking, the TGA has implemented a single email address (streamlinedsubmission@tga.gov.au) for the applicant through which formal correspondence will be coordinated. The TGA point of contact should be the PMAB Case Manager of the relevant clinical evaluation unit. Information on the areas of responsibility for the clinical units is included in Clinical evaluation sections for prescription medicines.
Pre-process activities
Pre-submission meetings
Applicants may wish to discuss scientific or procedural issues with the TGA in a pre-submission meeting prior to preparing a PPF or dossier. Such discussions should occur well in advance of PPF and dossier lodgement. The TGA strongly recommends that applicants organise a pre-submission meeting for complex applications. Meetings between TGA and applicants are conducted according to the processes described in the guideline Pre-submission meetings with the TGA. Applicants will need to provide details in the PPF and the dossier (Module 1.8.1) of any meetings conducted with the TGA.
Availability of dossier
The PPF requires applicants to identify a proposed dossier lodgement date. Applicants should not lodge a PPF until they are confident all the data necessary for the evaluation of the application will be ready for delivery on the proposed dossier lodgement date.
A dossier that was lodged previously with the TGA but not accepted for evaluation may be re-used in certain circumstances.
| Reason | Dossier can be reused? | Comments |
|---|---|---|
| Not accepted solely because dossier not lodged by required date | Yes - partially | The majority of the paper dossier can be re-used provided Modules 2 to 5 do not need to be updated or otherwise modified with new information. A revised paper copy of Module 1 must be provided. A complete electronic version of the revised dossier must be lodged. |
| Any other reason | No | The complete dossier must be re-lodged. The applicant retains responsibility for providing the TGA with the appropriate dossier to be evaluated. |
Pre-conditions
Applicants may need to fulfil a number of pre-conditions before lodging a PPF. In some cases, evidence is required in the PPF that the approval process has commenced on related activities. These are described below.
Orphan drug applications
For designated orphan drugs, the Secretary waives application and evaluation fees for the evaluation of a medicine for the purposes of registration. For fees to be waived, the applicant must apply for, and be granted by the TGA, orphan drug designation before lodging the PPF. Further information about making such a request can be found in:
Discussions with the TGA regarding orphan drug applications should commence well before the pre-submission phase. Applications for orphan drug designation should be lodged with the TGA two to three months before the intended start of the pre-submission phase. If an applicant has not received approval for the designation but chooses to proceed with the application, they become liable for the relevant application and evaluation fees.
Literature-based submissions
For applications using literature references as all or part of the supporting data set, the applicant must seek the TGA's agreement on the following before lodging the PPF:
- the search strategy employed
- the databases to be searched
- the criteria for determining which papers identified by the search results are to be included/excluded from the dossier.
These requirements are necessary because the data provided in the dossier is dependent on the search strategy. Applicants should plan to discuss these issues with the TGA at least three months prior to the pre-submission phase. Further information on literature-based submissions is provided in Literature-based submissions.
Fixed combination applications
For a new fixed combination product the applicant must justify, prior to lodging a PPF, the particular combination and the type and extent of data to be provided in the dossier. This is done by preparing and lodging with the TGA a 'justification for fixed combination'. This is a letter to the TGA that presents the case for the fixed combination, prepared according to the following EU guidelines that have been adopted in Australia:
- CHMP/EWP/240/95 Rev. 1 - Guideline on clinical development of fixed combination medicinal products
- EMEA/CHMP/SWP/258498/2005 - Guideline on the non-clinical development of fixed combinations of medicinal products
If one or more of the component active ingredients of the fixed combination product is/are not registered, the justification must include the proposed dossier lodgement dates for the unregistered components and the fixed combination product.
Ingredients
The TGA maintains databases of proprietary and non-proprietary ingredients which can be accessed from the TGA's eBS portal. Where an applicant wishes to register a new product that contains a new ingredient (one that is not on the databases), the applicant must commence, before lodging the PPF, the process for approval of a new ingredient for inclusion on the relevant databases.
For each new ingredient, the following forms must be lodged:
- a Notification of a New Proprietary Ingredient for each new proprietary ingredient
- an application form for proposing a new chemical name (AAN), biological name (ABN) and/or herbal name (AHN) for each new non-proprietary ingredient. See Forms for new ingredient names and proprietary ingredients.
Refer to part 2.1 of Information for applicants completing a pre-submission planning form for details on accessing the ingredient databases and the location of forms.
Note: Unless justified, toxicology data must be provided to the TGA to support the safety of an existing ingredient that is to be used for different purposes to that originally approved, for example, a new route of administration.
Genetically modified organisms (GMOs)
It is likely that approval from the Office of the Gene Technology Regulator (OGTR) is required for medicines that:
- contain GMO(s)
- consist of GMO(s)
- are manufactured in Australia and are derived from GMOs.
In such circumstances, applicants must apply to the OGTR for a licence, an exemption, or a Notifiable Low Risk Dealing approval, before lodging the PPF. Any OGTR advice (for example, OGTR licence, acknowledgement of receipt of licence application, other record of consent) must be provided to the TGA with the PPF.
Medicines scheduling
Where a proposed application relies upon the rescheduling of an existing substance in the Poisons Standard, applicants must lodge an application for rescheduling prior to lodging the PPF:
Concurrent applications
Applications to amend any details for a product cannot be lodged until the product is listed on the ARTG. If an applicant wishes to make changes to a proposed product, the application cannot be lodged until the registration process is finalised. The same requirement applies to a new chemical entity application currently under evaluation and a proposed new combination application (where the product contains the new chemical entity under evaluation).
Where a product is already registered and there is an existing application (variation or registration) currently with the TGA, it is acceptable to lodge a concurrent application in certain circumstances, provided that the changes proposed in the applications are unrelated. The following table indicates when it may be appropriate to lodge concurrent applications.
| Application currently with the TGA | ||||
|---|---|---|---|---|
| Resulting in new Register entry1 with new AUST R | Resulting in new Register entry1 but no new AUST R | Not resulting in a new Register entry1 | ||
| Proposed application | Resulting in new Register entry1 with new AUST R | Proposed application can be lodged only if new products covered in the proposed application are based on an existing Register entry. If not, proposed application should be postponed until application under evaluation has been approved and the ARTG updated. | Proposed application can be lodged only if the changes currently under evaluation are not to apply to the new products covered in the proposed application. | Proposed application should be postponed until application under evaluation has been approved and the ARTG updated. |
| Resulting in new Register entry1 but no new AUST R | Proposed application can be lodged only if changes are to apply to existing Register entries only. If not, proposed application should be postponed until application under evaluation has been approved and the ARTG updated. | Can be lodged, provided the changes in the current and proposed applications are unrelated. | Can be lodged, provided the changes in the current and proposed applications are unrelated. | |
| Not resulting in a new Register entry1 | Proposed application can be lodged only if changes are to apply to existing Register entries only. If not, proposed application should be postponed until application under evaluation has been approved and the ARTG updated. | Can be lodged, provided the changes in the current and proposed applications are unrelated. | Can be lodged, provided the changes in the current and proposed applications are unrelated. | |
1A new registration (or new Register entry) is one that requires a new ARTG entry by reason of being 'separate and distinct goods' under s.16 of the Therapeutic Goods Act 1989. This includes new chemical entities, new strengths, new dosage forms, different directions for use, formulation changes, changes in trade name, extensions of indication. However, not all new registrations will result in a new AUST R number being allocated if they are taken to be grouped by reason of the Therapeutic Goods (Groups) Order 2001.
If unsure of regulatory requirements where a related application is under consideration, applicants should contact the TGA before lodging a PPF for an application. Where there is a potential relationship between an application currently under evaluation and the proposed application, this must be indicated in the PPF.
Where the proposed application seeks changes to an entry in the Register under section 9D of the Act and would result in changes to the Product Information document currently under evaluation, such changes to the document currently under evaluation must be identified, either in the Product Information document or the letter of application of the proposed application.
In cases where a concurrent application is acceptable and an applicant chooses to lodge a concurrent application, the applicant assumes a level of risk. This is particularly so in cases where the outcome of the initial application is unknown.
It is possible that an application may be considered not effective if, for example:
- the outcome of the initial application is negative
- the evaluation on the initial application has not progressed sufficiently.
Registration process phases
-
Phase 1: Pre-submission
-
Phase 2: Submission
-
Phase 3: First round assessment
-
Phase 4: Consolidated section 31 request response
-
Phase 5: Second round assessment
-
Phase 6: Expert advisory review
-
Phase 7: Decision
-
Phase 8: Post-decision
Phase 1: Pre-submission phase
Flowchart showing pre-submission: phase 1 of registration process
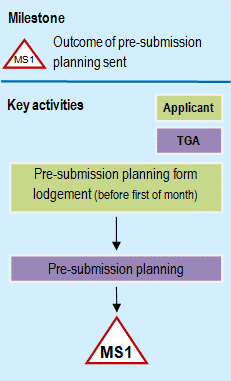
{kind=link}
A flowchart diagram showing pre-submission planning milestone MS1. At the top, there's a legend with a triangular MS1 symbol indicating 'Outcome of pre-submission planning sent'. The flowchart shows two parallel swim lanes labeled 'Applicant' (in light green) and 'TGA' (in purple). The process flows from top to bottom: starting with 'Pre-submission planning form lodgement' in the Applicant lane, flowing down to 'Pre-submission planning' in the TGA lane, and ending at the MS1 milestone marker. The diagram uses arrows to show the sequence of steps.
Objective
The pre-submission phase applies to category 1 and category 2 applications, excluding requests for additional trade names.
The pre-submission phase begins with the lodgement of the Pre-submission planning form (PPF).
The PPF provides the TGA with the necessary information on the scope and scale of an application to arrange appropriate resourcing for the processing and evaluation of the application.
Once a PPF has been considered complete and acceptable, the TGA begins the process of securing appropriate evaluators for the dossier.
A complete PPF identifies the proposed application type, and contains general information about the quality, nonclinical, and clinical evidence to be included in the dossier.
The information provided in the PPF allows the TGA to commit to timeframes for the evaluation of the application.
PPF-only pathway: questions and answers
Do I need to have an Australian Approved Name (AAN)/Australian Biological Name (ABN) approved by the TGA Naming Committee prior to lodging my submission under the PPF only pre-submission phase option?
Yes, as per standard registration process business rules, the requisite AAN or ABN should be approved before you lodge your submission.
The online application system requires a TGA approved AAN/ABN active ingredient name in order for you to enter formulation details and successfully lodge your application.
You should liaise with the TGA Naming Committee (TGAnames@tga.gov.au) as soon as you begin planning your submission and provide them with your planned lodgement timeframes.
Make sure that you have given the TGA Naming Committee sufficient lead time of about one month.
Do I need to lodge a Pre-submission Planning Form (PPF)?
Yes, it is still necessary to lodge a complete PPF. Make sure you meet all of the PPF requirements in relation to orphan drug designation, literature-based submission and new proprietary and non-proprietary ingredient names. For further information, refer to page 1 of the PPF.
Where can I find the correct PPF to use?
Complete the PPF which is accessible from the TGA website. The form must be completed in its entirety although there is no need to attach 'Module 2 or equivalent'. The Module 2 summaries will be required to be submitted with your dossier.
You may wish to attach additional documentation (for example, additional manufacturing site details if the existing form doesn't have enough space to include all of the proposed manufacturers).
Make sure sufficient detail about your application is provided, particularly in relation to the number and size of studies (eg, quality, non-clinical, clinical reports).
When can I lodge my PPF?
PPFs should be lodged to the TGA between the 1st and 15th day of the month. We encourage applicants to lodge their PPFs at the earliest possible date within the lodgement month.
When completing my pre-submission lodgement on eBS, what date should I select for 'You propose that the TGA commence validation of your submission dossier on'?
Select the earliest date available and we will commence validation of the submission dossier within the specified timeframe.
When completing my pre-submission lodgement on eBS, what date should I select for the 'Date on which your submission will arrive at TGA'?
To submit via the PPF-only pathway, the date on which your submission will arrive at TGA must be between then 16th of the month and the last day of the same month that your Pre-submission Planning Form (PPF) was lodged.
When will the TGA assign a stream number to complete my 'submission number'?
The TGA will continually monitor the PPFs as they enter the system and assign a stream number (based on your proposed indication) as soon as possible. This process completes your 'submission number' and allows you to finalise your eCTD dossier.
Use the complete ‘submission number’ (that includes the stream number) for all subsequent correspondence regarding the submission.
When can I lodge my dossier?
Submit your dossier via the usual procedure as soon as possible but no later than the end of the month in which you lodged your PPF.
Do I need to complete an application form?
Yes, make sure that your dossier is accompanied by the usual application form. Check the TGA website (Prescription medicine registration form) or contact the Application Entry Team (aet.application.entry.team@health.gov.au) for further information.
What applications are eligible for consideration?
The “PPF-only” pathway is available for all Category 1 prescription medicine application types.
Will I receive a Planning Letter and Evaluation Plan at Milestone 1?
No, there will be no formal Milestone 1. You should proceed to lodge your entire dossier as soon as the complete submission number is visible on eBS. This will occur once the TGA has added the relevant stream number based on your proposed indication (i.e. 'PM-yyyy-xxxxx-z-stream number'). Contact the Application Entry Team ((aet.application.entry.team@health.gov.au) if you experience any difficulties.
What happens if my PPF is lodged after the 15th of the month?
If your PPF is lodged onto the system after the 15th of the month cut-off, it may still be processed but may be batched with PPFs received during the following month's intake.
What happens if my dossier doesn't meet the Mandatory requirements for an effective application?
The dossiers will undergo electronic validation as well as administrative and technical screening as per the existing prescription medicines registration process. The same requirements must be met and the same decision-making principles will be applied. You will be notified as to whether your dossier has passed preliminary assessment at Milestone 2 through a Notification Letter.
If my application does not pass preliminary assessment, can a re-submitted application be considered under the PPF-only pre-submission phase option?
Yes, prior decisions made in relation to your product will not be taken into account when the eligibility criteria are applied.
What happens if my eCTD sequence doesn't pass TGA validation?
You will be notified as soon as possible, if your dossier does not pass validation. Depending on the validation issues, the submission may be deemed to have not passed preliminary assessment or processed with the next PPF batch.
To avoid this scenario, validate your dossiers prior to lodgement (via eSubmissions@health.gov.au) and contact the TGA immediately if you experience any difficulties (via your Case Manager at streamlinedsubmission@health.gov.au).
When will I receive an Evaluation Plan?
Should your application pass preliminary assessment, you will be issued with an Evaluation Plan at Milestone 2. An Evaluation Plan Estimator is available on the TGA website to facilitate your planning activities.
How do I complete Module 1.2.2, Module 1.7.2 and Module 1.7.3 in the dossier?
Include a PDF of your PPF in Module 1.2.2. Complete Modules 1.7.2 and 1.7.3 as you would under the existing registration process.
Milestone 1
If the PPF is considered complete and acceptable, a TGA Planning letter is sent to the applicant identifying the expected dossier lodgement date and target milestone dates for the application.
Key dates
The pre-submission phase commences with the lodgement of a PPF. PPFs are processed on the first day of each month.
The submission phase concludes when TGA sends the applicant a Planning letter. This letter is sent to applicants before or on the fifteenth of the month following the month in which the PPF was initially processed (or the following working day where the fifteenth of the month falls on a weekend or public holiday in the Australian Capital Territory).
Requirements at pre-submission
Pre-submission planning form
Applicants must complete and lodge a Pre-submission planning form. All sections of the PPF that are relevant to the proposed application must be completed. Information for applicants completing a pre-submission planning form is a step-by-step guide for applicants completing a PPF. If the applicant does not complete all relevant sections and provide the necessary documents as attachments, the TGA will consider the PPF not complete and not acceptable. Should the applicant wish to proceed with the registration of the medicine or the variation to the register entry, they will need to lodge a new PPF.
Module 2 data
A key element of the PPF is the provision of Module 2 (or equivalent) data which contains information on the scope, scale, and complexity of the proposed dossier. The TGA encourages the attachment of a complete CTD Module 2. The requirements for module 2 content at the pre-submission phase are explained in more detail in the Pre-submission planning form and Information for applicants completing a pre-submission planning form.
The TGA uses the information lodged by applicants in the PPF to plan the necessary resourcing to process and evaluate the application and to commit to milestone dates in the Planning letter. The information is particularly crucial when the TGA needs to secure external evaluators.
Lodging a pre-submission planning form
On-line form via eBS
Completion and lodgement of a PPF is a two-step process that uses the TGA's secure eBS portal (see Information for applicants completing a pre-submission planning form.)
Comment
The PPF is currently downloaded from the TGA website, completed, and lodged with the TGA through eBS. The TGA is working to develop an online smart form in eBS to replace this document. Smart form technology will allow the form to be pre-populated with automatically retrieved applicant information.
Certification by applicant
The applicant must declare on the PPF that they understand the TGA's requirements for an effective application and that the application will be processed by the TGA in accordance with the procedures described in this guidance.
The TGA's requirements for an effective application are established though legislative instruments made under section 9D and section 23 of the Act. Under these instruments, the planning information provided at pre-submission, along with the application form, and dossier (containing data for evaluation) comprise the information required under section 9D and section 23 of the Act to allow determination of the application.
Application fee
Upon lodgement of a PPF, applicants become liable to pay the application fee2. The application fee allows the TGA to recover the costs associated with processing the PPF and arranging evaluation resources. If a PPF is considered 'not effective' or the dossier is not subsequently accepted for evaluation, the applicant will forfeit this application fee. An application cannot be considered effective if the application fee has not been paid.
The application fee amounts are available at the fees and payments section of the TGA website.
Change of details after pre-submission
The TGA Planning letter (issued when a PPF is considered complete and acceptable) contains:
- the lodgement date for the dossier
- the expected dates for the milestones of the regulatory process
- any issues the TGA has identified when considering the PPF which need to be addressed in the dossier. For example, the TGA may indicate whether a proposed justification is not sound, giving the applicant time to make amendments before dossier lodgement.
When lodging the PPF, the applicant declares that they will provide a complete application dossier, the contents of which align with the information provided in the PPF, by the date that they have specified in the PPF. If it appears that the scope or scale of the dossier will be different to that described in the PPF, there is a risk that the application will be considered 'not effective'.
Examples of changes that are likely to result in an application being considered 'not effective' include:
- a significant change of proposed indication or addition of an indication
- an increase in the number of studies that form key sources of evidence (including, but not limited to, pivotal phase III studies, biopharmaceutic studies and carcinogenicity studies)
- a new strength
- an additional trade name.
The TGA recognises that some information may change before a dossier is lodged, for example, new safety information becomes available or further information needs to be included to address issues that the TGA raises in the Planning letter. Where the information that needs to be included impacts on the scope and scale of the application, applicants are advised to contact the TGA before lodging the application dossier.
Any differences between the dossier and the information provided in the PPF must be acknowledged in the covering letter for the dossier then described in detail and justified in Module 1.8.3: Declaration of compliance with the pre-submission planning form and planning letter. Refer to CTD Module 1 for more information on preparing these documents.
Applicants should note that where the application differs in scope or scale from that described in the PPF but the TGA agrees to proceed with evaluation, the TGA will need to reassess resources and may need to adjust milestone dates accordingly.
Application dates
The TGA Planning letter outlines the key dates for each phase of the regulatory process, including the expected date of dossier lodgement. The dossier may be lodged any time between the receipt of the Planning letter and close of business on the expected date of lodgement identified in the Planning letter. Applicants are encouraged to lodge their dossier as early as possible. However, early lodgement of the dossier will not result in an earlier achievement of milestone 2.
The Planning letter also identifies the date on which it is proposed to send the section 31 request for information or documents to the applicant and the date by which the applicant must respond to the request. This allows applicants to prepare to address any issues raised.
What happens if an applicant misses a key date?
To be included in a given monthly batch, the PPF must be received on or before the day preceding the first day of each month. A PPF lodged on the first day of a month will not be processed until the following month.
Phase 2: Submission phase
Flowchart showing submission: phase 2 of registration process
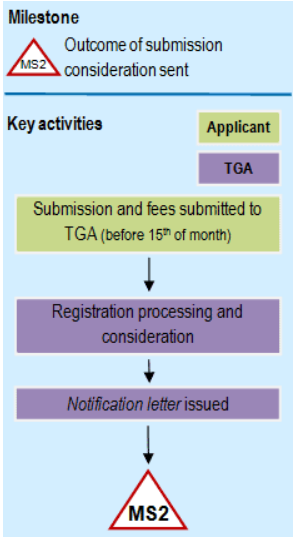
{kind=link}
Flow chart showing the 'MS2' milestone process for application submission and review. The diagram has two columns labeled 'Key activities' and 'Applicant/TGA' with color-coded boxes (green for Applicant, purple for TGA activities). The process flows from top to bottom:
- Starts with 'Submission and fees submitted to TGA (before 15th of month)' in green
- Flows to 'Registration processing and consideration' in purple
- Ends with 'Notification letter issued' in purple
- Concludes at an 'MS2' milestone marker (shown as a red triangle)
The title 'Milestone' appears at the top with text 'Outcome of submission consideration sent' under an MS2 triangle symbol.
Objective
The submission phase involves processing activities in preparation for application evaluation. For the TGA, processing activities include:
- confirmation of dossier delivery by the expected lodgement date
- verification that any application fee has been paid
- workflow planning and IT administration
- consideration of the application against the TGA regulatory requirements
- issuing of a Notification letter, including notice of evaluation fee payable, if applicable.
Milestone 2
The TGA sends a letter to the applicant identifying whether the application has been considered effective and accepted for evaluation, or considered 'not effective'.
Key dates
The submission phase commences with receipt of the dossier. The dossier is expected to contain all the information required by the TGA to evaluate the application(s).
For applications to register:
- a new chemical entity
- a new biological entity
- a new fixed combination
- a similar biological medicinal product
- an extension of indications
the dossier must be received by TGA before close of business (5.00 pm) on the seventh of the month following receipt of the Planning letter (or the following working day where the seventh falls on a weekend or public holiday in the Australian Capital Territory), unless another date is specified in the Planning letter.
For applications to:
- register a major variation
- register a minor variation
- register a new generic medicine
- vary the PI
the dossier must be received by TGA before close of business (5.00 pm) on the 14th of the month following receipt of the Planning letter (or the following working day where the fourteenth falls on a weekend or public holiday in the Australian Capital Territory).
The submission phase concludes when TGA sends the applicant a Notification letter. This letter is sent to applicants before the end of the month in which the dossier was lodged (or the following working day where the last day of the month falls on a weekend or public holiday in the Australian Capital Territory).
The Notification letter will advise the applicant whether the application is effective or not (Section 9D(7) and Section 23of the Act). If an application is considered not effective the Notification letter will explain why it was considered not effective.
Lodgement of application
Applicants of all new chemical entity, new fixed combination, and new generic medicine applications must electronically lodge their application through eBS prior to sending the hard and electronic copies of the dossier to the TGA. In this context, 'electronic lodgement' means the applicant has created an application in eBS and subsequently printed out an application form and an electronic lodgement cover sheet for inclusion in Module 1 of the dossier. Electronic lodgement, in this context, does not mean the dossier has been lodged electronically.
Processing the application
Effective applications
An application will be considered effective and accepted for evaluation if:
- the dossier arrives by the expected date (as identified in the Planning letter) following the lodgement of a complete PPF, and is consistent with the information in that form
- it includes the completed form: Application for the registration, or to vary the conditions of registration, of prescription medicines, or the eBS electronic equivalent (both of which include applicant declarations that all data for evaluation by the TGA has been presented at the time of dossier lodgement)
- it satisfies and is in accordance with relevant TGA regulatory requirements (see below).
TGA regulatory requirements
Applicants must abide by all applicable requirements. These requirements include, but are not limited to, those set out in:
- Therapeutic Goods Act 1989
- Therapeutic Goods Regulations 1990
- all relevant:
- Therapeutic Goods Orders
- ICH and CHMP guidelines that have been adopted by the TGA, and other Australia-specific guidelines
- legislative instruments made under section 9D and section 23 of the Act, which encompass:
- Pre-submission planning form
- Information for applicants completing a pre-submission planning form
- Application for the registration, or to vary the conditions of registration, of prescription medicines, or the eBS electronic equivalent
- CTD Module 1: Administrative information and prescribing information for Australia
- Common Technical Document (CTD)
- Mandatory requirements for an effective application
Evaluation fee
The full evaluation fee is due and payable when the applicant is notified that the application is accepted for evaluation. The Notification letter will specify the evaluation fee amount payable. If the evaluation fee for an application for the registration of a prescription medicine is not paid within two months from the date of the Notification letter, the application will lapse (see section 24(2) of the Act).
The evaluation fee amounts are available at the fees and payments section of the TGA website.
Applications not accepted
Applications that do not meet the TGA's regulatory requirements will be considered 'not effective'. Applicants of applications considered 'not effective' will be notified in writing of the reasons the application was not accepted for evaluation. If the applicant wishes to proceed with the application they must lodge a new PPF and potentially a new dossier (see section 4.2 - Availability of dossier).
The dossier becomes a Commonwealth record once processing commences and cannot be returned to the applicant.
What happens if an applicant misses a key date?
Applicants are encouraged to contact the TGA as soon as possible if they believe that they will be unable to lodge the dossier by the expected date to allow the TGA to reallocate resources that have been assigned to the application. A revised evaluation plan will be negotiated through the relevant Case Manager.
If an application does not arrive by the expected date, the TGA will reallocate the evaluation resources assigned to that application. In such cases, the applicant will forfeit any application fee and must begin the application process with lodgement of a new PPF. If processing of the application did not commence, an applicant will be able to make arrangements to retrieve the dossier.
Phase 3: First round assessment phase
Flowchart showing First round assessment: phase 3 of registration process

{kind=link}
A flowchart showing a milestone labeled MS3 for 'Outcome of first round assessment sent'. The diagram shows two participants: Applicant (in light green) and TGA (in purple). The process flows downward, starting with 'Assessment by primary and secondary evaluation units', followed by 'Consolidated section 31 request issued reports', and ending at a milestone marker labeled MS3. Arrows connect each step sequentially.
Objective
All data provided in the dossier are considered by the evaluators during the first round assessment phase. Where there are issues or questions about any component of the application, a consolidated section 31 request for information containing requests from all evaluation areas within the TGA is compiled and sent to the applicant by the date specified in the Planning letter.
Within this phase, TGA evaluators may directly contact the applicant to seek clarification or ask questions informally if they determine that waiting for the formal consolidated section 31 request on a minor clarification is unnecessary. This type of informal question will not change the timeline for the consolidated section 31 request.
Milestone 3
Applicants will be sent a Milestone 3 letter including:
- a consolidated section 31 request for information or documents, if required
- copies of the first round assessment reports prepared by the quality, nonclinical, clinical and RMP evaluators.
Key dates
The first round assessment phase commences the day after the Notification letter has been sent.
The first round assessment phase concludes 4 (generic applications) or 5 (all other applications) months later when TGA sends the applicant a Milestone 3 letter. This letter is sent to applicants before the end of the applicable month (or the following working day where the last day of the month falls on a weekend or public holiday in the Australian Capital Territory).
Assessment of data
Period for evaluation
The default timeframes for the first round evaluation of the data are:
- 3 months for the evaluation of new generic medicine applications
- 4 months for the evaluation of other applications.
Following evaluation of the data, a consolidated section 31 request for information or documents will be compiled, checked, and authorised within 1 month of the completion of the first round assessment and sent to the applicant. This allows the TGA to ensure that issues and concerns from all evaluation areas are clearly identified and consolidated into the request.
External evaluators
The TGA may contract external evaluators to review aspects of the dossier. The TGA will coordinate the evaluation with the external evaluator as part of the first round assessment phase. Communication with the applicant in relation to an evaluation will always be through the TGA.
First round assessment reports
The first round assessment reports are sent with the Milestone 3 letter and will help to provide a context for any request for information or documents.
Phase 4: Consolidated section 31 request response phase
Flowchart showing Phase 4: Consolidated section 31 request response phase
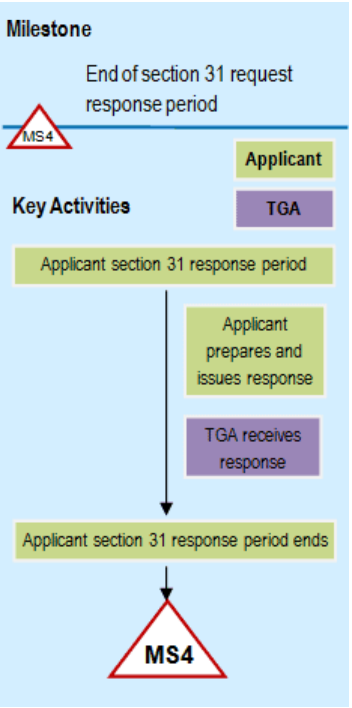
{kind=link}
A flowchart showing the milestone process for a Section 31 request response period.
The diagram has a light blue background and consists of two main sections labelled 'Milestone' and 'Key Activities'. At the top, a milestone marker labelled 'MS4' indicates 'End of section 31 request response period'.
The key activities flow vertically downward, color-coded for two parties: Applicant (in light green boxes) and TGA (in purple boxes).
The sequence shows: 'Applicant section 31 response period', followed by two parallel activities - 'Applicant prepares and issues response' and 'TGA receives response'. The flow concludes with 'Applicant section 31 response period ends' leading to a final milestone marker 'MS4'.
A legend indicates that light green boxes represent Applicant activities and purple boxes represent TGA activities.
Objective
The consolidated section 31 request response phase allows applicants time to consider the TGA's consolidated section 31 request for information or documents, prepare a response and send the response to the TGA.
Milestone 4
Applicants will send TGA a response to:
- any consolidated section 31 request for information or documents
- the first round assessment reports prepared by the quality, nonclinical and clinical evaluators, if required.
Key dates
The consolidated section 31 request response phase commences on completion of the first round assessment phase when the TGA sends the consolidated section 31 request for information or documents to the applicant.
The consolidated section 31 request response phase ends on the date by which the applicant is required to respond to the section 31 as identified in the section 31 request letter. The section 31 response time will be either 30 or 60 days as nominated by applicant in the PPF and confirmed by the TGA in the Planning letter.
Note: The consolidated section 31 request response period does not count towards the statutory 255 working day period for evaluation (Regulation 16A(2)).
Response to section 31 request
Responses to a section 31 request for information or documents must be provided in CTD format. Applicants need to send both hard copy and electronic copy formats of the response to the TGA. Information on providing a section 31 response is contained in CTD Module 1: Administrative information and prescribing information for Australia.
Applicants should pay close attention to the questions raised in the s31 letter, as well as any requests in the letter for other information or documents, as this may be the sole opportunity that an applicant has to provide this information.
Response to first round assessment reports
Applicants should review the first round assessment reports and advise the TGA of any perceived errors of fact or major omissions. Each identified error of fact or omission must be referenced to information previously submitted (Study number and location, for example, Module 3.2.P.4.3 Method validation, p.23). It is not appropriate to submit new data to negate a perceived error of fact or omission (see Section 2.5 Information). Although there is no limit to the size of an applicant's response, it is expected that the response to any one report should be no more than six single sided A4 pages and in a font no smaller than 12 point.
Responses to the assessment reports must be included in Module 1.0.2. and should be submitted with, but as separate file(s) to, the response to the section 31 request. For those applications where there is no section 31 request, applicants must respond by the date specified in the Milestone 3 letter. At least 14 calendar days of TGA issuing the first round assessment reports will be provided.
What happens if an applicant misses a key date?
The consolidated section 31 request response phase will end on the due date specified in the section 31 letter irrespective of whether an applicant's response to the request is received by the TGA.
Should the applicant not reply in the nominated time period, the second round assessment phase will be concluded based on the information available to the TGA.
Phase 5: Second round assessment phase
Flowchart showing phase 5: Second round assessment phase
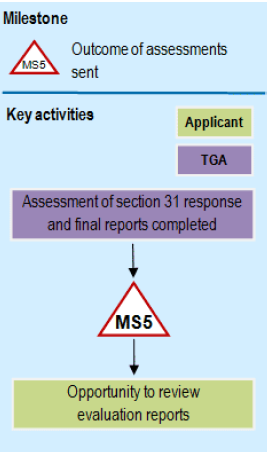
{kind=link}
A flowchart showing milestone MS5 process.
The diagram flows vertically with two parallel swim lanes labeled 'TGA' in purple and 'Applicant' in light green.
The process starts with 'Assessment of section 31 response and final reports completed' in the TGA lane, flowing down to a milestone marker labelled 'MS5'.
This leads to an 'Opportunity to review evaluation reports' step in the Applicant lane. At the top of the diagram is a legend indicating that MS5 represents 'Outcome of assessments sent'.
Objective
During the second round assessment phase, evaluators will consider the response provided by the applicant to the section 31 request (if applicable) and complete the evaluation of the data.
Milestone 5
All second round assessment reports are completed.
Key dates
The second round assessment phase will commence on completion of the consolidated section 31 request response phase, whether or not a response is received from the applicant.
The TGA will complete the second round assessment reports:
- within two months for applications for new generic medicines
- within one month for all other applications.
TGA will issue the second round assessment reports at the completion of the second round assessment phase.
Assessment of response
During the second round assessment phase evaluators consider the applicant's section 31 response to questions raised in relation to issues identified in the first round assessment. Where the section 31 response is inadequate or raises further issues, evaluators may liaise directly with applicants for clarification, but no additional time will be added and no additional section 31 requests will be formally sent. Any outstanding issues will be highlighted in the evaluation reports. These may be significant enough to warrant a recommendation to reject the application or they may be minor, possibly leading to the imposition of conditions of registration for applicant compliance.
The final reports are independent assessments of the data that include advice for consideration by the decision delegate.
The TGA has no way of anticipating at the pre-submission phase whether a consolidated section 31 request will be needed or how many questions or requests for documents might be contained in such a request. Unless the TGA contacts the applicant, applicants should expect that the milestone dates identified in the Planning letter will be retained even if there is no section 31 request.
Opportunity to review evaluation reports
Where the application is not referred to the Advisory Committee, applicants will be given a period of 14 calendar days after the TGA issues the final evaluation report(s) in which to review and advise the TGA of any perceived errors of fact or major omissions.
Where the application is referred to the Advisory Committee, comments in relation to perceived errors of fact or major omissions in the second round assessment reports should be received by the TGA at least 13 working days before the meeting date.
Phase 6: Expert advisory review phase
Flowchart showing Phase 6: Expert advisory review phase

{kind=link}
Flow chart showing the advisory committee process. The chart begins with a triangle labelled 'MS4' at the top and ends with a triangle labelled 'MS6' at the bottom. The chart is divided into two columns: 'Key activities' and 'Applicant/TGA', with TGA activities shown in purple boxes and Applicant activities in green.
The process flows vertically through these steps:
- Delegate's request for ACM/ACV advice
- Pre-ACM/ACV response and response to second round assessment reports
- ACM/ACV papers circulated
- ACM/ACV meeting
- ACM/ACV advice prepared and issued
Each step is connected by downward-pointing arrows, creating a linear workflow from top to bottom. The chart illustrates the formal sequence of activities between the applicant and TGA during the advisory committee process.
Objective
After completion of the second round assessment phase, the evaluation reports are considered by the delegate. The delegate may seek independent advice on issues concerning the application.
The main advisory group for prescription medicines generally is the Advisory Committee on Medicines (ACM) and for vaccines specifically it is the Advisory Committee on Vaccines (ACV). Independent expert advice may also be sought by the delegate outside of the advisory committees.
Information on each of the advisory committees is available on the TGA website, including proposed meeting dates.
Milestone 6
The TGA notifies the applicant of the advice received from the ACM or ACV.
Key dates
The expert advisory review phase commences on the day the delegate's Request for ACM/ACV advice (also referred to as the Delegate's Overview for ACM/ACV) is sent to the applicant. This date is at least 23 working days prior to the proposed ACM/ACV meeting.
The Pre-ACM/ACV response from the applicant must be is received by the committee secretariat at least 13 working days before the meeting date. This is to ensure that ACM/ACV have adequate time to consider the information.
The expert advisory review phase ends when the ACM/ACV advice is sent to the applicant. This generally occurs on the Friday two weeks after the meeting (10 working days after the ACM meeting or 12 working days after the ACV meeting).
Advisory Committee on Medicines and Advisory Committee on Vaccines
Request for ACM/ACV advice
The delegate will review the evaluation reports and prepare a Request for ACM advice or Request for ACV advice if independent advice from the ACM/ACV is required on aspects of the application.
The delegate is not obligated to refer an application to an expert advisory committee. At the time of preparing the Planning letter it may not always be clear whether an application will require ACM/ACV advice; however, for applications other than those to register a new generic medicine, the Planning letter will identify the ACM/ACV meeting allocated to the application should the delegate decide to refer aspects of the application for advice.
When preparing the Request for ACM advice, the delegate will consider any outstanding issues from the evaluation process. The request contains a number of elements including:
- an overview of the application including key findings from the evaluation reports
- outstanding issues identified by the delegate
- an account of the risks and benefits relevant to the proposed product and proposed indication(s)
- a specific request for advice on any aspect of the application's characteristics, findings, or risk/benefit considerations
- whether special registration conditions, PI changes, or risk management plan changes would be appropriate.
On completion, a copy of the request is sent to the applicant prior to the ACM/ACV meeting to allow the applicant to compile a Pre-ACM/ACV response.
The delegate may also provide a Delegate's Overview for applications that do not require ACM/ACV advice. The purpose of this document is to summarise the evaluation and request information from the sponsor on any outstanding issues. This document will be clearly identified as not requiring ACM/ACV advice and therefore a Pre-ACM/ACV response will not be required. However a response addressing any outstanding evaluation issues may be required.
Pre-ACM/ACV response
The Pre-ACM/ACV response is the applicant's opportunity to provide comments that will be considered by the ACM/ACV. Applicants generally compile a Pre-ACM/ACV response based on the evaluation reports and the delegate's Request for ACM/ACV advice. The applicant should ensure they address any outstanding issues raised by the TGA, but should not provide new unsolicited information.
Guidelines for compiling the Pre ACM/ACV response
The response must be provided:
- as an electronic submission:
- via email as a zipped attachment (maximum size 20 megabytes), or
- on electronic media
- as an appropriately validated sequence using either the eCTD or NeeS specification, consistent with preceding sequence
- as sequence type 'Supplementary Information'; sequence description 'Pre-Advisory Committee response'
- as PDF files generated from electronically-sourced documents without placeholder documents. Include only the components that are necessary for the submission:
- if using eCTD specification, include only ‘new’ documents (that is, not previously submitted in an earlier sequence)
- if using NeeS specification, include a complete set of documents, even if previously submitted in an earlier sequence.
with a cover letter, containing an address block, including:
Sponsor's name [or consultant's name] (as appropriate)
Return address
Attention: Regulatory affairs officer's name
Email address
Phone number
Regarding the format, the response must:
- be clearly marked 'PRE-ACM RESPONSE' or 'PRE-ACV RESPONSE' in bold, large upper case type at the top of the first page
- be no more than ten pages in a font no smaller than 12 point
- clearly identify upfront any changes to the indications and/or dosage and administration information from the original application.
The following should be included (as separate files) with the response:
- A tabulation of any serious unexpected adverse drug reactions which are not mentioned in the proposed Australian Product Information and have not been submitted previously - it is expected that this would normally not exceed six pages. (include under Module 1.0.3 Response to request for information).
- The updated proposed Australian Product Information and the most up-to-date Consumer Medicine Information (CMI). Highlight changes from the version submitted with your application and reference the source of the change (e.g. clinical evaluation, company base document) in the margin. Ensure that you comply with Form for providing Product Information (Module 1.3.1 Product Information and Module 1.3.2 Consumer Medicine Information).
- statement on the current international regulatory status of the medicine. This should detail approvals (with indications), deferrals, withdrawals and rejections (including reasons). (Module 1.11.1 Foreign regulatory status).
- Where available, provide foreign equivalent to Product Information (in Module 1.11.2 Foreign Product Information)
- A copy of the latest Periodic Safety Update Report (PSUR), unless already supplied or none is available. (Module 5.3.6 Reports of Post-Marketing Experience)
The following table provides a guide to the location of the documents in CTD format:
| Pre-ACM/ACV component | CTD location* |
|---|---|
| Pre- ACM/ACV covering letter | Module 1.0.1 Cover letter |
| - | Module 1.0.2 Lifecycle management tracking table |
| Pre-ACM/ACV response | Module 1.0.3 Response to request for information |
| International regulatory history | Module 1.11.1 Foreign regulatory status |
| Adverse reactions update | Module 1.0.3 Response to request for information# |
| Sponsor's comments on PI | Module 1.0.3 Response to request for information# |
| Sponsor's comments on foreign PI | Module 1.0.3 Response to request for information# |
| PSUR | Module 5.3.6 Reports of Post-Marketing Experience |
| PI (annotated and non-annotated) | Module 1.3.1.1 Product Information - clean |
| Module 1.3.1.2 Product Information - annotated | |
| CMI (annotated and non-annotated) | Module 1.3.2.1 Consumer Medicine Information - clean |
| Module 1.3.2.2 Consumer Medicine Information - annotated | |
| Foreign equivalent(s) to Product Information | Module 1.11.2 Foreign Product Information# |
# Where multiple documents are submitted in the same CTD Module, clearly identify the different documents.
More information on Pre-ACM responses is available from the ACM Secretariat at ACM@health.gov.au, and for Pre-ACV responses from the ACV Secretariat at ACV@health.gov.au.
Withdrawing a submission: AusPAR requirements
The sponsor may withdraw the submission at any time during the expert advisory review phase.
When the withdrawal letter is received by the TGA will determine whether an AusPAR is required. Refer to the AusPAR guidance for information on AusPAR business rules.
Outcome of the ACM/ACV meeting
The ACM/ACV advice is documented in the ACM/ACV 'minutes' document, which is provided to the delegate and applicant following the meeting. This generally occurs on the Friday two weeks after the meeting (10 working days after the ACM meeting or 12 working days after the ACV meeting).
The ACM/ACV advice is independent and is primarily based on their expert opinion. Note that the ACM and ACV do not make decisions and their advice does not constitute directions to the Secretary or the TGA delegate.
The ACM/ACV advice in relation to the specific questions put to the ACM/ACV is published in the Australian public assessment report (AusPAR). The ACM/ACV minutes may also be provided to a number of overseas regulatory agencies as permitted under section 61(4) of the Act.
The ACM/ACV minutes, advice and agenda papers may also be provided to persons, bodies and authorities as permitted under section 61(5AA) of the Act.
What happens if an applicant misses a key date?
Where the applicant cannot provide a pre-ACM/ACV response to the ACM/ACV secretariat at least two weeks prior to the meeting date, the response will generally not be tabled for consideration by the ACM/ACV, but will be considered in the decision phase by the TGA delegate. Early contact with the committee secretariat and the case manager is advised if delays are expected.
Phase 7: Decision phase
Flowchart showing phase 7: Decision phase

{kind=link}
A flowchart showing a milestone process labelled 'MS7: Decision made by delegate'.
The chart flows vertically with two parallel swim lanes labelled 'Key Activities' and 'Applicant'. Under Key Activities, there are three connected purple boxes: 'Delegate's review and decision' flows down to 'Decision letter prepared and issued', ending at a red triangle labelled 'MS7'.
The Applicant lane contains a yellow box labelled 'TGA'. The flowchart uses arrows to show the sequence of steps.
Objective
The TGA delegate will determine whether the application is to be approved (possibly modified or varied) or rejected. Where any outstanding issues may affect the decision, the delegate may liaise directly with the applicant during this phase before finalising the decision.
For applications made under section 23 of the Act the applicant will be notified in writing of the decision within 28 days of it being made (Section 25(4) of the Act). The issuing of a decision ends the legislated evaluation period.
Milestone 7
A decision is made for approval or rejection of the application for a new registration or for a variation to a registration and the decision letter is sent to the applicant.
Key dates
For applications referred to the ACM/ACV for advice:
- the decision phase will commence when the ACM/ACV advice is sent to the applicant 10 working days after the Advisory Committee meeting
- the decision phase is completed when the applicant is notified of the delegate's decision 6 weeks after the ACM/ACV meeting.
For applications for new generic medicines:
- the decision phase will commence when the second round assessment reports, prepared by the relevant evaluators, are sent to the applicant
- the decision phase is completed when the applicant is notified of the delegate's decision 6 weeks after the second round assessment reports are sent to the applicant.
For other applications:
- the decision phase will commence when the second round assessment reports, prepared by the quality, nonclinical, clinical and RMP evaluators, are sent to the applicant
- the decision phase is completed when the applicant is notified of the delegate's decision 6 weeks after the ACM/ACV meeting that would have considered the application, if the application had been considered by the ACM/ACV.
Review and decision
Applicant's opportunity to review evaluation reports
Applicants will be given a period of 14 calendar days after the TGA issues the final evaluation report(s) in which to review and advise the TGA of any perceived errors of fact or major omissions.
Each identified error of fact or omission must be referenced to information previously submitted (Study number and location, for example, Module 3.2.P.4.3 Method validation, p.23). It is not appropriate to submit new data to negate a perceived error of fact or omission (see Section 2.5 Information). Although there is no limit to the size of an applicant's response, it is expected that the response to any one report should be no more than six single sided A4 pages and in a font no smaller than 12 point.
Responses to the assessment reports must be included in Module 1.0.2.
Applicants should also use this period to revise the draft PI based on recommendations in the evaluation reports. Identify clearly on any revised documents:
- changes to the documents which accord fully with the recommendations of the evaluator
- changes which are partially in accord with the evaluator's recommendations, together with reasons for not fully adopting them (for example, by cross-referring to data or to the response outlining omissions or errors of fact in the evaluation)
- reasons for contesting the evaluator's recommendations
- new amendments, analogous to safety related requests, affecting safety matters.
Revised PI documents must be included in Module 1.3.1.
Delegate's review
The delegate reviews all documentation associated with the application and makes an assessment of the risks and benefits. The review may address a number of outstanding issues including suggestions for revision of the Product Information (PI) or risk management plan (RMP). It may also address outstanding general registration details. The delegate may negotiate issues with the applicant prior to making a decision.
Decision for approval
Where the delegate is satisfied that benefits sufficiently outweigh risks associated with a product, the delegate will send the applicant a decision letter for approval of a new registration (possibly varied or modified) or approval to vary a registration. There may be special conditions of registration associated with the approval; these may include the conduct of post-market activities by the applicant, or a revision of existing documentation. The decision letter will also set out the appeal rights.
Decision for rejection
If a delegate decides to reject an application, a formal statement of the reasons is included in the decision letter. In addition to the reasons for the decision, the letter will set out the appeal rights.
In cases where an ACM or ACV recommendation to reject an application is unexpected the delegate will usually contact the applicant to discuss the proposed actions.
What happens if an applicant misses a key date?
There is no obligation for an applicant to provide a response to the evaluation reports. If no response is received by TGA within the 14 calendar days period after the TGA issues the final evaluation report(s), the delegate will proceed to make a decision based on the available information.
Applicants are responsible for monitoring existing GMP clearances that may be due to expire prior to or during the decision phase. A delegate is unable the approve an application for a new registration unless:
- all Australian manufacturer(s) involved hold a valid licence for the relevant production steps
- all overseas manufacturer(s) involved hold a valid GMP clearance for relevant production steps.
Phase 8: Post-decision phase
Flowchart showing phase 8: post-decision phase
{kind=link}
A flowchart diagram showing a project milestone labeled MS8. At the top, there's a triangle symbol containing 'MS8' with text stating 'Administrative and regulatory activities complete'.
Below this are 'Key activities' with a color-coded legend showing 'Applicant' in yellow-green and 'TGA' in purple.
The main flow shows a purple box labelled 'Final administration' with an arrow pointing down to another triangular MS8 symbol at the bottom. The image uses a light blue background.
Objective
During the post-decision phase, administrative and regulatory activities are completed.
Milestone 8
Administrative and regulatory activities are completed. Any outstanding payments are finalised (if applicable) and the ARTG entry is finalised.
Key dates
The post-decision phase commences when the applicant is notified of the delegate's decision.
The post-decision phase is completed by the end of the month following the delegate's decision.
Note: If an appeal is lodged prior to milestone 8, the milestone will be delayed until all appeal activities are complete. Under section 60 of the Act, an appeal by the applicant must be lodged within 90 days of notification of the decision.
ARTG registration
A new Register entry3 can only be included on the ARTG once the applicant provides a patent certificate or notice (section 26B(1) of the Act) to the TGA (see section 25(4) of the Act).
The TGA will check that the provisional ARTG record (originally provided by the applicant at dossier lodgement and updated during evaluation) matches the approved product details. The provisional ARTG record will become the ARTG Record of Registration.
The applicant must notify the TGA of the actual date of marketing commencement and meet other conditions of registration outlined in the delegate's decision letter.
Medicines information
After registration medicines information about the product and the decision process will be made available to the public through the TGA website. This information will be published as:
an Australian Public Assessment Report (AusPAR)
The AusPAR provides information about the evaluation of a prescription medicine and the considerations that led the TGA to approve or not approve an application. The AusPAR is drafted during the post-decision phase and sent to the applicant for review prior to publication at About Australian Public Assessment Reports for prescription medicines (AusPARs). Details of the contents, rules, and activities associated with an AusPAR are provided in the Understanding Australian Public Assessment Reports (AusPARs) for prescription medicines guidance.
an Australian Prescription Medicine Decision Summary
The Australian Prescription Medicine Decision Summary provides a short overview of the TGA's evaluation process leading to the registration of a new prescription medicine on the ARTG. Australian Prescription Medicine Decision Summaries are drafted during the post-decision phase and published at Australian prescription medicine decision summaries.
Product Information (PI)
Product Information document(s) in the approved form must be lodged by the applicant with the TGA within 2 weeks of the date of registration of the product(s).
Consumer Medicine Information (CMI)
CMI document(s) must be lodged with the TGA:
- for new product(s) - prior to first supply of the product(s)
- for variations to existing products - within 2 weeks of the date of approval of the variation.
What happens if an applicant misses a key date?
The post-decision phase for new register entries cannot be completed unless the section 26B(1) Certificate required in relation to patents is provided by the applicant.
Appendix 1: Sample timelines
| Activity | Date | Milestone | |
|---|---|---|---|
| Pre-submission phase | |||
| Applicant submits pre-submission planning form | 30 Nov 2012 | ||
| TGA commences processing PPF | 1 Dec 2012(3 Dec 2012*) | ||
| TGA Planning letter issued | On/before 15 Jan 2013 | MS1 | |
| Submission phase | |||
| Applicant lodges dossier | On/before 7 Feb 2013 | ||
| TGA Notification letter issued | On/before 28 Feb 2013 | MS2 | |
| First round assessment phase | |||
| Commencement of evaluation | 1 Mar 2013 | Planned evaluation time is 10.5 months | |
| Consolidated section 31 request sent from TGA | On/before 31 Jul 2013 | MS3 | |
| Consolidated section 31 request response period | |||
| 30 day option - selected at PPF | On/before 30 Aug 2013 | MS4 | |
| 60 day option - selected at PPF | On/before 29 Sep 2013 (30 Sep 2013*) | MS4 | |
| Second round assessment phase (assuming 30 day response period) | |||
| Completion of second round assessment | On/before 30 Sep 2013 | MS5 | |
| Expert advisory review phase (assuming submission referred) | |||
| Request for ACM advice sent to applicant | 5 Nov 2013 | ||
| Pre-ACM response received | 19 Nov 2013 | ||
| ACM meeting | 6 Dec 2013 | ||
| ACM advice finalised | 20 Dec 2013 (23 Dec 2013*) | MS6 | |
| Decision phase | |||
| Decision date | On/before 15 Jan 2014 | MS7 | |
| Post-decision phase | |||
| ARTG entry created | On/before 15 Feb 2014 (17 Feb 2014*) | MS8 | |
*Where the target date falls on a weekend or public holiday, the actual target date will be the next working day.
| Activity | Date | Milestone | |
|---|---|---|---|
| Pre-submission phase | |||
| Applicant submits pre-submission planning form | 31 Oct 2012 | ||
| TGA commences processing PPF | 1 Nov 2012 | ||
| TGA Planning letter issued | On/before 15 Dec 2012 (17 Dec 2012*) | MS1 | |
| Submission phase | |||
| Applicant lodges dossier | On/before 15 Jan 2013 | ||
| TGA Notification letter issued | On/before 31 Jan 2013 | MS2 | |
| First round assessment phase | |||
| Commencement of evaluation | 1 Feb 2013 | Planned evaluation time is 8.5 months | |
| Consolidated section 31 request sent from TGA | On/before 31 May 2013 | MS3 | |
| Consolidated section 31 request response period | |||
| 30 day option - selected at PPF | On/before 30 Jun 2013 (1 Jul 2013*) | MS4 | |
| 60 day option - selected at PPF | On/before 30 Jul 2013 | MS4 | |
| Second round assessment phase (assuming 30 day response period) | |||
| Completion of second round assessment | On/before 31 Aug 2013 (2 Sep 2013*) | MS5 | |
| Decision phase | |||
| Decision date | On/before 15 Oct 2013 | MS7 | |
| Post-decision phase | |||
| ARTG entry created | On/before 15 Nov 2013 | MS8 | |
*Where the target date falls on a weekend or public holiday, the actual target date will be the next working day.
Appendix 2: Registration process regulatory phases
The following diagram shows the phases and milestones of the registration process.
Glossary
| Term | Definition |
|---|---|
| Application | Refers to the regulatory activity required in respect of a product (a specific set of formulations, strengths and presentations) as requested by the applicant of the product. It is the specific set of information on the product submitted for review. Examples include:
|
| Application category | Identified by a number, for example, category 1 and category 2 applications, and refers to the overall legislated timeframes for decisions about applications. |
| Application type | Relates to the fees associated with an application and is identified by a letter, for example, A, B or C application types. Examples include:
|
| Dossier | Contains and/or references the necessary data to demonstrate the quality, safety and efficacy of a prescription medicine. The supporting data consists of:
|
| Submission | A collection of one or more applications that are grouped together for fee purposes, and is specific to a particular active ingredient and applicant. |
Footnotes
- A new registration (or new Register entry) is one that requires a new ARTG entry by reason of being 'separate and distinct goods' under s.16 of the Therapeutic Goods Act 1989. This includes new chemical entities, new strengths, new dosage forms, different directions for use, formulation changes, changes in trade name, extensions of indication. However, not all new registrations will result in a new AUST R number being allocated if they are taken to be grouped by reason of the Therapeutic Goods (Groups) Order 2001.
- An application fee is not payable for applications made under section 23 of the Act and involving a medicine which has been designated an orphan drug.
- A new registration (or new Register entry) is one that requires a new ARTG entry by reason of being 'separate and distinct goods' under s.16 of the Therapeutic Goods Act 1989. This includes new chemical entities, new strengths, new dosage forms, different directions for use, formulation changes, changes in trade name, and extensions of indication. However, not all new registrations will result in the allocation of a new AUST R number if they are taken to be grouped by reason of the Therapeutic Goods (Groups) Order 2001.
Downloads
Page history
Updates to expert advisory review phase
- Use of the phrase ACM/ACV rather than only ACM
- Provide additional details regarding the Pre-ACM/ACV responses (within 5.6.4.2)
- Update timeframes for provision of ACM/ACV minutes in line with current practice (within 5.6.3 & 5.6.4.4)
- Removal of the Pharmaceutical subcommittee information
- Expectation for reviewing evaluation reports combined for both applications that attend ACM/ACV and that do not attend ACM/ACV (within 5.7.4.1)
- Inclusion of information about Decision Summaries (within 5.8.5)
- Minor edits to increase readability.
Minor updates to Submissions.
Updated to reflect Provisional approval pathway.
Update to reflect new legislation.
Addition of link to guidance on orphan drug designations.
Second version to reflect outcomes from the public and stakeholder consultation, the introduction of a separate workflow for generic medicines and the opportunity for applicants to consider all evaluation reports at milestone 3.
Original publication.
Updates to expert advisory review phase
- Use of the phrase ACM/ACV rather than only ACM
- Provide additional details regarding the Pre-ACM/ACV responses (within 5.6.4.2)
- Update timeframes for provision of ACM/ACV minutes in line with current practice (within 5.6.3 & 5.6.4.4)
- Removal of the Pharmaceutical subcommittee information
- Expectation for reviewing evaluation reports combined for both applications that attend ACM/ACV and that do not attend ACM/ACV (within 5.7.4.1)
- Inclusion of information about Decision Summaries (within 5.8.5)
- Minor edits to increase readability.
Minor updates to Submissions.
Updated to reflect Provisional approval pathway.
Update to reflect new legislation.
Addition of link to guidance on orphan drug designations.
Second version to reflect outcomes from the public and stakeholder consultation, the introduction of a separate workflow for generic medicines and the opportunity for applicants to consider all evaluation reports at milestone 3.
Original publication.