We’re experiencing issues for some mobile devices with our ARTG dataset filter function. We apologise for any inconvenience while we work to fix this.
Purpose
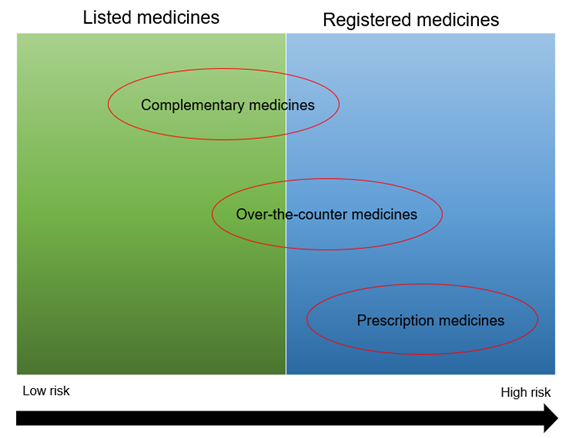
This guidance is intended for consumers, health professionals and industry. It provides a general overview of the Australian legislative framework for low risk (listed) medicines and registered complementary medicines. It explains the different types of medicines and the different entry pathways into the Australian Register of Therapeutic Goods (ARTG). It also provides information on legislation, medicine ingredients, medicine interface issues, practitioner and product exemptions, supplying medicines and post market monitoring.
{kind=link}