Including in-vitro diagnostic (IVD) medical devices in the ARTG
Guidance for sponsors on the process for including IVD medical devices in the ARTG.
Purpose
This guidance outlines the requirements and processes for including in-vitro diagnostic medical devices (IVDs) in the Australian Register of Therapeutic Goods (ARTG), which is essential for the import, supply, and export of devices in Australia. The information covers various classes of IVDs, sponsor responsibilities, compliance requirements with Essential Principles, and conformity assessment procedures, along with notable exceptions where ARTG inclusion is not required.
Including IVD medical devices in the ARTG
Overview
The ARTG is a register of therapeutic goods accepted for importation into Australia, supply for use in Australia, or exportation from Australia.
Medical devices1 cannot generally be imported, supplied in or exported from Australia unless they are included in the ARTG.
Exceptions to this requirement include devices that are supplied through one of the following mechanisms for supplying medical devices in Australia not included in the ARTG:
- Clinical Trials in Australia
- Authorised Prescribers
- Special Access Scheme
- Personal Importation
- Exemptions in the national interest, to deal with emergencies
For more information on these mechanisms please see the Australian Regulatory Guidelines for Medical Devices (ARGMD) or refer to Parts 4-6A and 4-7 of the Therapeutic Goods Act 1989 (the Act) and Schedule 4 of the Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations).
Class 1-3 in-house IVD medical devices are required to be entered on an in-house IVD database that is separate from the ARTG.
Only an Australian sponsor can apply to include an IVD in the ARTG. For more information, please see our guidance on Understanding conformity assessment for in-vitro diagnostic medical devices (IVDs): for sponsors.
A sponsor can apply to include an IVD in the ARTG if:
- the device complies with the Essential Principles; and
- an appropriate conformity assessment procedure has been applied to the device.
Other requirements that must be complied with are outlined in this Section.
All inclusions in the ARTG are subject to automatic conditions and further conditions may be imposed by the TGA where it is appropriate.
Depending on the type of IVD to be included in the ARTG, slightly different processes need to be followed. These processes are for:
- IVDs intended for export only
- Class 1 IVDs
- IVDs other than Class 1
Process for including export only IVD medical devices in the ARTG
IVDs intended for export only are either manufactured in Australia solely for export or are imported into Australia for the purposes of export. Export only IVDs need to be included in the ARTG as Class 1 IVDs and cannot be supplied to users in Australia. An IVD that is imported solely for the purpose of export is exempt from the requirement to be included in the ARTG, provided it remains subject to the control of the Australian Customs Service and is not subject to any actions that could be considered an act of manufacturing in Australia.
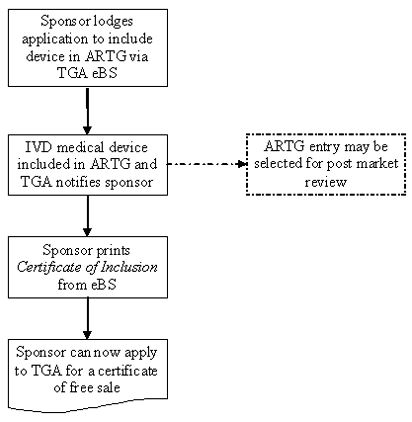
The following flowchart summarises the steps for including an export only IVD in the ARTG:
Process for including export only IVD medical devices in the ARTG
{kind=link}
This image shows a vertical flowchart depicting the process for including an IVD medical device in the Australian Register of Therapeutic Goods (ARTG). The flowchart consists of five boxes connected by arrows, with one box being optional (shown with dotted lines).
The process flows as follows:
- Start: "Sponsor lodges application to include device in ARTG via TGA eBS"
- Next step: "IVD medical device included in ARTG and TGA notifies sponsor"
- Optional step (connected with dotted line): "ARTG entry may be selected for post market review"
- Next step: "Sponsor prints Certificate of Inclusion from eBS"
- Final step: "Sponsor can now apply to TGA for a certificate of free sale"
All boxes are connected by single downward-pointing arrows, except for the optional post-market review box, which is connected by a dotted line horizontally to the second step. The text is presented in a clear, readable font, and the flowchart uses a simple black-and-white colour scheme.
Process for including Class 1 IVD medical devices (other than export only) in the ARTG
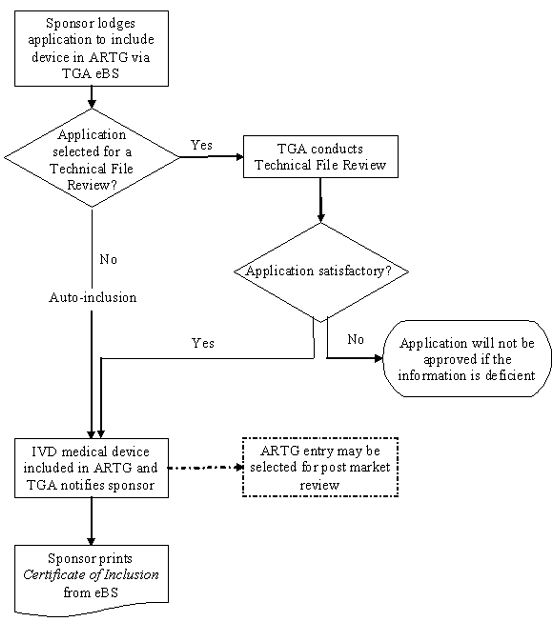
The following flowchart summarises the steps for including a Class 1 IVD that is to be supplied in Australia in the ARTG:
Process for including Class 1 IVD medical devices (other than export only) in the ARTG
{kind=link}
Flowchart showing the process for including a device in ARTG (Australian Register of Therapeutic Goods). Process starts with sponsor lodging application via TGA eBS. Path splits based on Technical File Review selection. If selected, TGA reviews for satisfactory completion. If satisfactory or if auto-included, device is included in ARTG and sponsor is notified. Sponsor then prints Certificate of Inclusion. A dotted box indicates possible post-market review. Applications with deficient information are not approved.
Process for including IVD medical devices (other than Class 1) in the ARTG
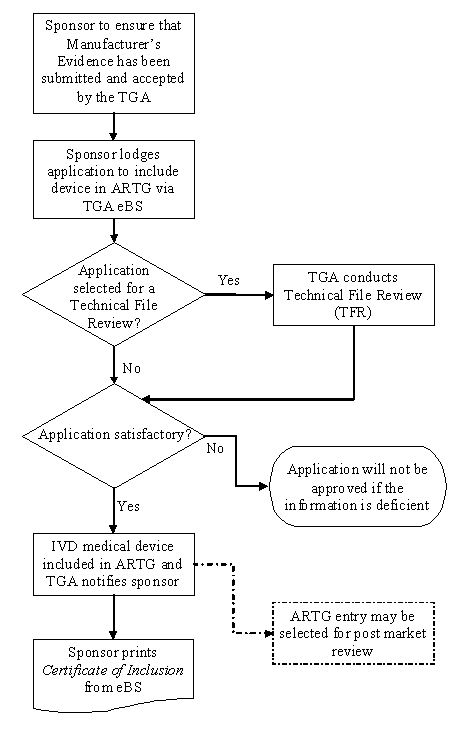
The following flowchart summarises the steps for including an IVD that is to be supplied in Australia, other than a Class 1 IVD, in the ARTG:
Process for including an IVD that is to be supplied in Australia, other than a Class 1 IVD, in the ARTG
{kind=link}
Flowchart shows the TGA application process for medical devices.
Steps include:
- Manufacturer's Evidence submission and acceptance
- Sponsor's ARTG application via TGA eBS
- Decision point for Technical File Review
- Application assessment for satisfactory completion.
If approved, the IVD device is included in ARTG and sponsor receives notification, followed by Certificate printing. Deficient applications are rejected. A dotted line indicates possible post-market review. Process uses diamond shapes for decisions and rectangles for actions.
Applications for inclusion in the ARTG
The Australian sponsor must lodge an application to include a device or devices in the ARTG using eBS.
The manufacturer's quality management system certificate or conformity assessment certificate, which may also be referred to as Manufacturer's Evidence, is not required for Class 1 IVDs. Manufacturers of Class 1 IVDs must still apply the appropriate conformity assessment procedure and prepare an Australian Declaration of Conformity (DOC), this needs to be submitted to the TGA with the device application. The sponsor must provide evidence of conformity to the TGA upon request.
Manufacturer's Evidence is required for all Class 2, 3 and 4 IVDs. Before lodging an application, sponsors must submit and receive notification that the manufacturer's quality management systems certification or conformity assessment certificate has been accepted by the TGA. For more information on acceptable manufacturer's evidence and its submission please see our Understanding conformity assessment for in-vitro diagnostic medical devices (IVDs): for manufacturers guidance.
The process for lodging an application for inclusion in the ARTG requires the sponsor to undertake the following, in accordance with Section 41FC of the Act:
- complete the appropriate application form
- submit the completed application to the TGA
- pay the prescribed application fee
- ensure that, if a conformity assessment certificate is required for the device, the appropriate certificate has been obtained before lodging the application for inclusion of the IVD in the ARTG
- ensure that the application does not contain information that is false or misleading.
The following must also be provided with the application lodged through eBS:
- Australian Declaration of Conformity for Class 3 IVDs
- TGA-issued Design Examination certificate or Type Examination certificate, as appropriate, for Class 4 IVDs
If the application is for a Class 3 IVD and a CMDCAS ISO 13485 certificate has been selected to support IVDs in the application, evidence of the Class 3 Canadian Medical Device Licence for the IVDs supported by the CMDCAS ISO 13485 certificate, if an active licence exists, should also be provided.
When lodging an application, the sponsor must certify in accordance with Section 41FD of the Act that:
- the device/s are medical devices
- the device/s are intended for a specified purpose
- the device/s are correctly classified according to the medical device classifications
- the device/s comply with the Essential Principles
- they have:
- available sufficient information to substantiate compliance with the Essential Principles, or
- procedures in place, including a written agreement with the manufacturer of the device/s to ensure that this information can be obtained from the manufacturer within the period required by the TGA
- an appropriate conformity assessment procedure has been applied to the device/s
- they have:
- available sufficient information to substantiate the application of those conformity assessment procedures, or
- procedures in place, including a written agreement with the manufacturer of the device/ to ensure that this information can be obtained from the manufacturer within the period required by the TGA
- the device/s comply with every requirement (if any) relating to advertising
- the device/s do not contain substances that are prohibited imports for the purposes of the Customs Act 1901
- the information included in or with the application is complete and correct.
There are criminal and civil penalties for making false statements in connection with a 41FD certification or an application for inclusion in the ARTG.
For Class 1 IVDs, most successful applications lodged in eBS will result in an 'automatic' inclusion in the ARTG. This means that there will be no further assessment of the application by the TGA prior to the device being included in the ARTG.
However, some devices in applications for Class 1, 2 and 3 IVDs may be subject to a mandatory or non-mandatory application audit which involves checking some or all aspects of the application and certifications. Application audits that are conducted on IVDs are generally limited to the product technical file and are often referred to by the TGA as Technical File Reviews (TFRs).
Section 41FH of the Therapeutic Goods Act 1989 (the Act) specifies that:
- applications to include certain medical devices in the ARTG must be selected for an Application Audit and an assessment fee will be charged. If however, the evidence of conformity submitted prior to lodging the application is a TGA Conformity Assessment Certificate, an audit will not be conducted as the necessary assessments are considered to have already been conducted.
- the TGA may select any other applications for inclusion to undergo an Application Audit - an assessment fee will not be charged for these non-mandatory audits.
For more information on Application Audits see - Application audits of IVD medical device applications.
In summary:
| If | then | and |
|---|---|---|
| an application to include a device in the ARTG is successful | the TGA will notify the sponsor that the application has been successful and | the sponsor can print the Certificate of Inclusion on eBS. |
| an application to include a device in the ARTG is not successful | the TGA will notify the sponsor in writing that the application has not been successful | the sponsor should ensure that they have addressed any deficiencies in the information provided to the TGA before they re-apply. |
Kinds of IVD medical devices
An inclusion in the ARTG is for a kind of medical device. This means that an entry in the ARTG may cover a range of products that are of the same kind rather than individual devices.
From the Therapeutic Goods Act 1989 - 41BE Kinds of medical devices
- For the purposes of this Chapter, a medical device is taken to be of the same kind as another medical device if they:
- have the same sponsor; and
- have the same manufacturer; and
- have the same device nomenclature system code (see subsection (3)); and
- have the same medical device classification; and
- are the same in relation to such other characteristics as the regulations prescribe, either generally or in relation to medical devices of the kind in question.
From the Therapeutic Goods (Medical Devices) Regulations 2002 - 1.6 Kinds of medical devices - other common characteristics
For paragraph 41BE (1) (e) of the Act, in relation to any of the following medical devices, a characteristic is the unique product identifier given to the device by its manufacturer to identify the device and any variants:
- a Class 4 IVD medical device, other than an immunohaematology reagent IVD medical device that is a Class 4 IVD medical device
In the case of Class 1, Class 2, Class 3 IVDs and Class 4 immunohaematology reagents (IHRs), one IVD is considered to be of the "same kind" as another IVD, if both devices:
- have the same legal manufacturer and
- have the same sponsor and
- are the same classification and
- have the same GMDN code.
Provided these criteria are met, a single entry in the ARTG may encompass multiple devices. There is no record kept in the ARTG of the product family name, product numbers, or catalogue numbers for Class 1, Class 2 or Class 3 IVD medical devices. However, for Class 4 IHRs and Class 1-3 IVDs that have undergone a mandatory technical file review, the names of the individual IVDs covered by an entry will appear in the ARTG. If a sponsor wishes to add a new IVD to an existing entry for a "kind of medical device", and the new IVD is subject to a mandatory technical file review, an application for variation to the ARTG needs to be submitted.
For Class 4 IVDs (including Class 4 in-house IVDs) that are not IHRs, a further requirement is added to the definition of same kind of medical device - they must have the same Unique Product Identifier (UPI).
How to group Class 1, Class 2 or Class 3 IVDs
Described below is an example of a "kind of medical device" that is a Class 2 IVD:
Manufacturer "ABC Pty Ltd" produces a range of kits, calibrators, controls and test reagents intended to be used for the measurement of a number of different proteins in clinical specimens. The range of tests includes total protein, albumin, C-reactive protein (CRP), ferritin, transferrin and also a number of different classes of immunoglobulins. The manufacturer has classified each of the products as a Class 2 IVD.
Sponsor "XYZ Pty Ltd" wishes to import the full range of assays measuring protein-based analytes and supply them in Australia. Before the sponsor imports the IVDs, they obtain the manufacturer's Australian Declaration of Conformity and establish that the manufacturer has classified each IVD as a Class 2 IVD, and that the same GMDN collective term, "CT974 Clinical chemistry-specific protein IVDs" has been applied to each. The sponsor then confirms that each test within the range of IVDs for the measurement of different proteins therefore has:
- the same manufacturer (ABC Pty Ltd)
- the same risk classification (Class 2 IVD)
- the same GMDN code (CT974 Clinical chemistry-specific protein IVDs).
Because each of the different tests to be imported fit the parameters that describe an IVD "of the same kind", there is no requirement for multiple ARTG entries, even though the individual kits may have different trade names and detect different proteins (e.g. "ABC Blue C-reactive protein kit", "ABC Red ferritin kit" etc.). The unique name for each product is not considered within the definition for a "kind of medical device" for Class 1-3 IVDs. Therefore, sponsor XYZ Pty Ltd submits an application to the TGA to include the full range of Clinical chemistry-specific protein IVDs under a single entry in the ARTG.
Sponsor "QRS Pty Ltd" also wishes to supply the same range of assays for the measurement of the different proteins to laboratories across Australia, and has arranged to source the kits from manufacturer "ABC Pty Ltd". They discover that ABC Pty Ltd is already entered in the ARTG as the manufacturer of these produces, however because QRS Pty Ltd is not the same sponsor as identified in the existing ARTG entry, they will need to apply to the TGA to have the same range of Clinical chemistry-specific protein IVDs included in the ARTG under their name before they may import them for use in Australia.
This scenario highlights that when two different sponsors import the same products for use in Australia, they are regarded as different "kinds of medical devices" and therefore each sponsor is required to have their own separate ARTG entries. This requirement only applies to sponsors, and not to users or marketing agents who obtain their products from the sponsor for distribution or on-sale.
New IVDs can be taken to be covered by an existing inclusion provided that they are the same "kind of medical device" as described in Section 41BE of the Act. However, if the device is of a kind that would be selected for an application audit for any of the reasons listed in Regulation 5.3 Applications to be selected for auditing, a variation must be submitted and approved before the device can be considered to be covered by an inclusion.
How to group Class 3 or Class 4 Immunohaematology reagents (IHRs)
Described below is an example of a "kind of medical device" that is an Immunohaematology reagent (IHR).
Sponsor "XYZ Pty Ltd" also wishes to import blood grouping antisera manufactured by "ABC Pty Ltd" for use in testing the ABO and Rhesus blood group systems. The manufacturer has classified each individual reagent as Class 4 IVD and applied the GMDN code "CT887 Immunohaematology blood grouping antisera IVDs". The IHRs therefore have:
- The same manufacturer (ABC Pty Ltd)
- The same classification (Class 4 IVD)
- The same GMDN code (CT887 Immunohaematology blood grouping antisera IVDs).
Because each of the blood grouping antisera does not result in a change to any of the parameters describing a "kind of medical device", they do not require separate ARTG entries, even though they detect markers from different blood group systems. Therefore, an application can be made to include the antisera in a single entry in the ARTG.
Typically, a manufacturer will produce a range of different antisera and may also produce typed reagent red blood cells for use as IHRs. A product range will commonly encompass both Class 3 and Class 4 IVDs. If for example, ABC Pty Ltd manufactured antisera for the Lutheran blood group system (a Class 3 IVD) as well as ABO and Rhesus blood grouping antisera (Class 4 IVDs) then the entire range could not be grouped as the same "kind of medical device" in a single ARTG entry because all the antisera are not assigned the same risk classification.
Under the classification rules, IHRs can only be classified as either Class 3 or Class 4 IVDs, therefore when considering a range of IHRs there can be a maximum of two entries per GMDN collective term code (i.e. one for Class 3 and one for Class 4 IVDs), provided they also have:
- the same manufacturer; and
- the same sponsor.
Unique Product Identifiers (UPIs)
The UPI is the combination of words, numbers, symbols or letters assigned by the manufacturer to uniquely identify an individual IVD, or a combination of IVDs which together constitute an IVD closed system.
An IVD closed system is a combination of reagents, calibrators and quality control materials that:
- share a common intended purpose; and
- are to be used only in combination with each other as components of a single assay.
Closed systems are usually dedicated for use with a single instrument.
The UPI is generally different from the catalogue or stock unit identifier assigned to the device and usually the family name, assay name and/or test kit name will form a hierarchy in identifying the device.
Different manufacturers identify their product lines in different ways such as:
- using family names to identify a range of products that use similar platform technology;
- uniquely identifying each device with an assay name;
- specifying the technology or method an IVD uses;
- specifying the disease marker a device tests for; or
- a combination of these approaches.
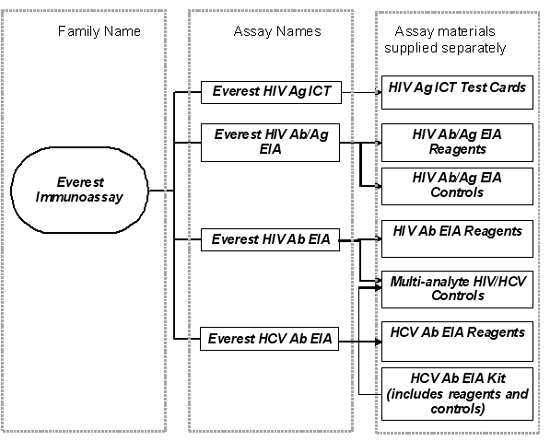
For example, a family of immunoassays may be represented as follows:
Everest Immunoassay product family structure
{kind=link}
Organisational chart with three columns: Family Name, Assay Names, and Assay Materials. The Everest Immunoassay family includes four main assays: Everest HIV Ag ICT (with test cards), Everest HIV Ab/Ag EIA (with reagents and controls), Everest HIV Ab EIA (with reagents and multi-analyte HIV/HCV controls), and Everest HCV Ab EIA (with reagents and complete kit). Each assay connects to its corresponding materials shown in the rightmost column. Columns are separated by dotted borders.
In this example, the family name "Everest Immunoassay" is not considered a UPI, because it does not distinguish between the individual IVDs and the closed systems in the product range - i.e. HIV Ag ICT Test Cards vs. HIV Ab/Ag EIA.
The HIV Ab/Ag EIA Reagents and HIV Ab/Ag EIA Controls together form an IVD closed system since the reagents and controls are dedicated for use only with each other. The UPI for this closed system would therefore be Everest HIV Ab/Ag EIA.
Separately supplied components of the Everest HIV Ab EIA and Everest HCV Ab EIA assays cannot be regarded as a closed system because they share the same multi-analyte controls. The multi-analyte controls may be used with the HIV Ab EIA Reagents, the HCV Ab EIA Reagents and they may also be used with the HCV Ab EIA Kit as an alternate to controls supplied in the kit. Constituents of a closed system must be used only in combination with each other as components of a single assay with a common intended purpose.
In the example given above, the following kit names are considered UPIs:
- Everest HIV Ag ICT Test Cards
- Everest HIV Ab/Ag EIA
- Everest HIV Ab EIA Reagents
- Everest Multi-analyte HIV/HCV Controls
- Everest HCV Ab EIA Reagents
- Everest HCV Ab EIA Kit (includes reagents and controls).
Please note: Different pack sizes, for example a 100 test kit or a 500 test kit of the same IVD, are not considered to form part of the UPI.
Conditions on inclusion in the ARTG
All inclusions of IVD medical devices in the ARTG are subject to conditions. There are:
- automatic conditions, which are imposed at the time a device is included in the ARTG;
- other (additional) conditions, which may be imposed by the TGA when device/s are included in the ARTG; and
- conditions imposed after devices are included in the ARTG.
Automatic conditions on inclusion in the ARTG
In accordance with section 41FN of the Act, the following conditions on inclusion apply automatically:
| Type of condition | Description |
|---|---|
| Entry and inspection powers | An authorised person be allowed to:
|
| Delivery for samples | If requested by the TGA, the sponsor will deliver a reasonable number of samples of a device |
| Availability of information about a device | The TGA may request information at any time while a device is included in the ARTG:
The sponsor must have procedures in place, including a written agreement with the manufacturer of the devices, to ensure that information required by the Regulations can be obtained from the manufacturer within 20 working days. The sponsor must also report adverse events to the TGA within specified mandatory timeframes and assist in their investigation. |
| Advertising materials | Advertising material relating to the medical device is consistent with the intended purpose as certified in the application for inclusion in the ARTG. |
Conditions which may be imposed on an inclusion in the ARTG
In accordance with section 41FO of the Act, the TGA may impose additional conditions when including the kind of device in the ARTG. These conditions may be imposed to address any specific concerns regarding the manufacture, storage or disposal of products, keeping records and tracking devices, or any other issues relating to quality, safety and performance.
Conditions imposed after devices are included in the ARTG
In accordance with section 41FP of the Act, the TGA may:
- impose new conditions on the kind of device included in the ARTG
- vary or remove existing conditions.
Imposing a new condition, or the variation or removal of an existing condition takes effect:
- if the notice states that the action is necessary to prevent imminent risk of death, serious illness or serious injury - on the day on which the notice is given to the person
- in any other case - on the day specified in the notice, not earlier than 20 working days after the notice is given to the sponsor.
Certificates of Inclusion
Sponsors will be notified by the TGA if their application for inclusion in the ARTG has been successful. The notification will include instructions for printing the Certificate of Inclusion from eBS.
Applications for amendments to entries in the ARTG
If a sponsor needs to amend the details of an IVD that is already included in the ARTG, they should access eBS and complete the appropriate form. For more information on changes to entries in the ARTG please see our Vary medical device entries in the ARTG (includes IVDs) webpage.
Footnotes
- The use of the term 'medical device' is taken to include 'IVD medical devices or IVDs' - the latter terms are used to denote references that are specific to IVD medical devices.
Page history
Title changed from 'Including IVD medical devices in the ARTG' to 'Including in-vitro diagnostic (IVD) medical devices in the ARTG' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updates following Class I Validation project – export only devices
Original publication
Title changed from 'Including IVD medical devices in the ARTG' to 'Including in-vitro diagnostic (IVD) medical devices in the ARTG' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updates following Class I Validation project – export only devices
Original publication