Procedure for recalls, product alerts and product corrections (PRAC)
Information for sponsors conducting market actions for therapeutic goods in Australia.
The new Procedure for Recalls, Product Alerts and Product Corrections (PRAC), took effect on 5 March 2025.
The PRAC replaces the Uniform Recall Procedure for Therapeutic Goods (URPTG) as the procedure for sponsors to follow when conducting market actions in Australia.
What’s changed from the URPTG to the PRAC?
While the PRAC is a complete re-write of the URPTG to improve its content and readability, the process for sponsors performing a market action largely remains the same.
Some of the changes introduced in the PRAC include:
- updating the recall terminology, removing the sometimes confusing categories of ‘recall’ and ‘non-recall’ actions, and replacing them with one category called ‘market actions’
- condensing the document and reducing the number of steps in the recall process from 10 to 5
- placing information in easy to digest formats including tables or pictures, where appropriate
- clarifying when we require extra information for actions which may create a shortage, or involve lengthy corrections, etc
- simplifying the definitions for Class and Level of market actions without changing their meanings
- removing information concerning our legislative powers related to recalls. This information will be updated and placed as a separate guidance piece on the TGA website when the PRAC takes effect
- new processes where we will:
- distribute sponsors’ approved customer letters with our TGA notification to the state and territory recall coordinators
- no longer verify the accuracy of sponsor customer lists through protracted back-and-forth dialogues. Sponsors are expected to submit accurate information to avoid delays.
The PRAC retains the new reforms introduced in the final version of the URPTG (Version 2.4) in March 2024, including flexible reporting requirements, greater transparency around the 'Early Advice' process, and new user-friendly templates.
Immediate actions
If your problem relates to any of the following
- Imminent and significant risks to patient lives or public health
- Actual or suspected tampering
- Radiopharmaceuticals
- Blood or blood components
- Biological or human tissue
- Clinical trials
You should do the following
- Immediately instruct your customers to quarantine affected goods
- To prevent further use or supply, confirm that your customers have:
- notified impacted surgeons or clinicians (where applicable)
- notified other areas who may have received the goods
- Then contact us (details below) promptly and be prepared to follow our instructions
Contact us as soon as possible if you’re unsure whether to take an immediate action. For instance, if the action could create a shortage or disrupt critical patient care, etc.
Purpose
This resource guides sponsors (‘you’) on performing market actions to address problems with therapeutic goods supplied to customers that may pose a risk to public health and safety.
This page references TGA templates which are freely available for download.
What we mean by ‘problem’
We use ‘problem’ to refer to situations that impact any of the following:
- safety
- quality
- efficacy (medicines and biologicals)
- performance (medical devices)
- presentation
- use of the product.
Non-compliance to standards, manufacturing deviations, contamination, and customer misuse are examples of problems that could prompt a market action.
What we mean by ‘end user’
We refer to ‘end users’ as those who use the product or have it used on them, such as doctors or patients.
Contact details
| recalls@health.gov.au | |
| Phone |
Users who are deaf or have a hearing or speech impairment can call through the National Relay Service:
|
Contact details for TGA, State and Territory Recall coordinators.
Undertaking an action
To protect public health and safety, all market actions must:
- be undertaken by the sponsor or the person responsible for supplying the goods
- follow this procedure in consultation with us
- involve all those who are affected by the problem.
Delegates of the Secretary of the Australian Government Department of Health and Aged Care can exercise powers under the Therapeutic Goods Act 1989 (the Act) to compel you to perform market actions for therapeutic goods, if necessary.
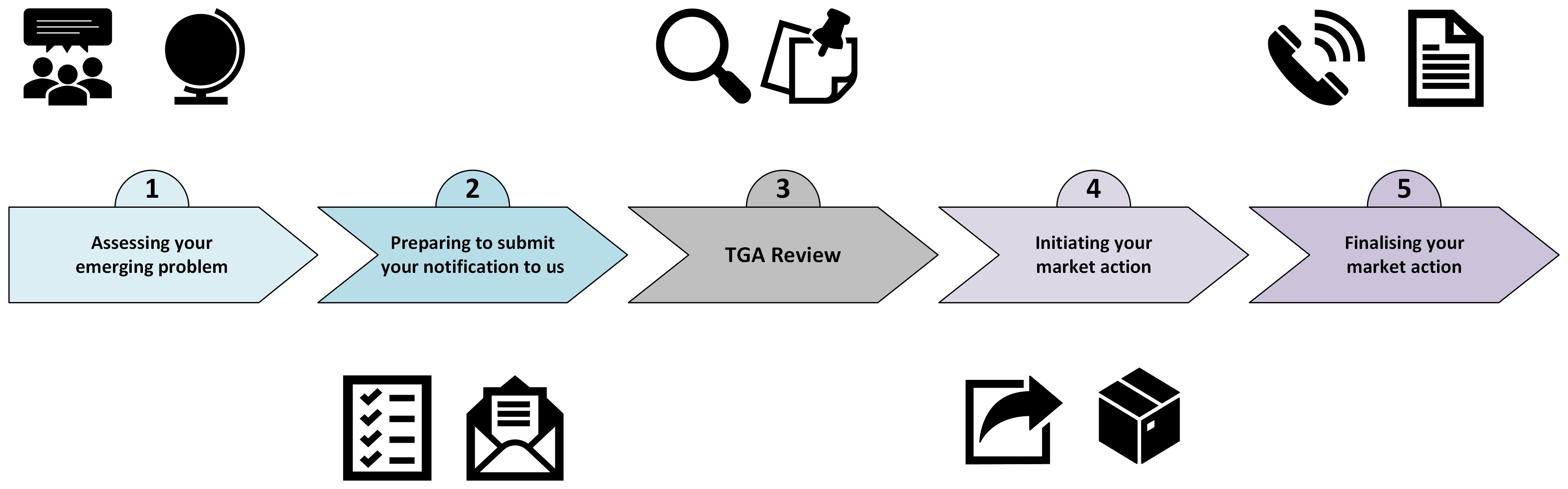
The end-to-end market action process
Follow steps 1 to 5 to successfully complete your market action.
{kind=link}
- Assessing your emerging problem
- Preparing to submit your notification to us
- TGA review
- Initiating your market action
- Finalising your market action
Step 1: Assessing your emerging problem
You should gather as much information about your problem as possible, to ensure that we can assess your notification promptly once submitted. For safety-related problems, do not delay in notifying us even if you are missing some details - we would rather be aware of emerging problems, especially ones with significant safety risks.
Details of affected goods
You should know, where applicable:
| Product identifiers | Quantity and distribution | Problem details |
|---|---|---|
|
|
|
You should also know the following details for specific types of products:
| Medicines | Medical devices | Bloods and biologicals |
|---|---|---|
|
|
|
Risk analyses
When a manufacturer notifies you of a problem or defect, you should receive their risk analysis (also called health risk assessments (HRA), health hazard evaluations (HHE), etc.). Ensure that you are aware of the conclusions of your manufacturer’s risk assessment when submitting it to us.
If you do not have the manufacturer’s risk analysis for any reason (for example, if the problem has arisen from local complaints and/or adverse events), you should:
- gather as much information as you can
- detail the complaints and/or adverse events to the manufacturer and request a risk analysis from them.
What the risk analysis should include
We will follow up if the following is not addressed:
| Details of the problem | Hazard severity | Manufacturer's proposed corrective action |
|---|---|---|
|
|
|
Commercially sensitive or personal information
You must call attention to commercially sensitive information in your notification.
Do not give us any personal information or private details of any patients/individuals.
We will manage any information that is commercially sensitive or private in line with the TGA’s privacy policy.
Step 2: Preparing and submitting your notification to us
What is a notification?
Your ‘notification’ explains the problem and your proposed mitigation. It consists of a completed online form and supporting documents submitted through the TGA eBusiness Services portal (Business Services > Applications > Market actions). The online form contains a Help guide, hover text and examples for your convenience.
Documents you must include

{kind=link}
Documents to include:
- Risk assessment
- Customer list
- Customer letter and response form
- Risk assessment (root cause, probability and severity)
- Customer list (all affected customers you have supplied and will contact, in the following format: State, Customer Name, Suburb)
- Customer letter (what you will send your customers)
- Customer response form (which customers will use to confirm they have received and read your letter)
You must attach these under the ‘Supporting Information’ tab within the online form.
Unless otherwise permitted, do not contact your customers until you receive our agreement to do so after our review (Step 3).
Accessing TGA eBusiness Services (TBS)
If you have queries about the TBS portal, or do not have access;
- refer to our guide on getting started with the TGA, or;
- contact the TBS Helpdesk: ebs@tga.gov.au or 1800 010 624.
TGA templates
Our TGA templates are presented in a ‘fill-in-the-blanks’ style showing what information should be included, and how. We strongly encourage the use of our templates to ensure that you include all required information.
Regarding the accuracy of customer lists
We no longer verify the accuracy of sponsor’s customer lists. We will now only remove the names of private individuals before distribution in accordance with our privacy policy.
When we provide market action information to the state and territory recall coordinators and other key stakeholders, we instruct them to follow up with sponsors directly regarding the quality of the customer information submitted.
To avoid delays, sponsors are expected to submit accurate information in the required format using the correct TGA template.
Critical information
Our review requires that we understand your action strategy and potential consequences. You must tell us if:
- You and/or your suppliers cannot identify all end users
- You will have difficulty recovering or correcting the goods
- You anticipate:
- Supply shortages/disruptions to critical patient care
- Adverse events before you can recover/correct the goods
- The action could take longer than 3 months to complete.
Situations requiring additional documents
If you or your suppliers cannot identify all end users (e.g. distribution via retailers), or if we determine the problem is likely to become ‘high profile’ (e.g. may attract public or media attention) – we may require you to write and submit a completed public notice, which you will also distribute (in Step 4) via:
- Your website
- Retail stores
- Print media advertisements
- Social media or digital posts or
- Targeted methods (e.g: SMS alerts).
We will work with you to determine the most appropriate place(s) to distribute this notice.
Electronic response forms
You may use a QR code / link to an online survey response form to allow customers to respond to you electronically. Your QR code / electronic form should:
- be linked under the header ‘response form’ in your customer letter
- align with the text of our response form template
- be working when you submit your notification so we can review the text of the link or electronic response form.
If you need to update your customer list or letter
If you discover after submitting your notification that additional customers have been impacted by the problem, or that another action is more appropriate, contact us and provide the updated documents.
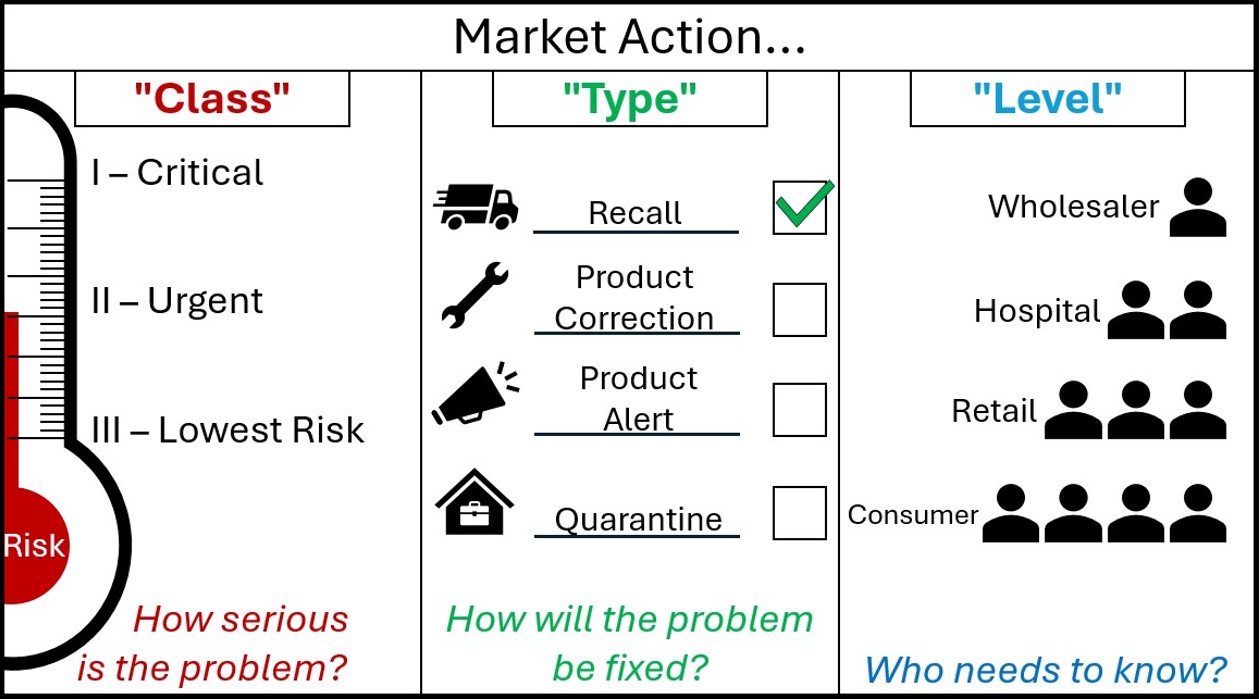
Market action nomenclature
Market actions are defined across 3 categories.
{kind=link}
A graphic with the header 'Market Action'.
The graphic is split into 3 segments.
The first segment is marked 'Class - How serious is the Problem?'
It shows market action class as a thermometer marked 'Risk'. As the risk increases on the thermometer, the Class goes from Three - 'Lowest Risk', to Two 'Urgent', to One - 'Critical'.
The Second Graphic is marked 'Type - How will the problem be fixed?'.
It shows market action type as four options, each with an associated image. A speeding truck for the Recall action type, a spanner for the Product Correction Action type, a megaphone for the Product Alert Action type, and a locked box within a house for the Quarantine Action Type.
Next to each action are tickboxes, showing that these are choices that sponsors can select. For decoration purposes, the Recall Action type is ticked.
The Third Graphic is marked 'Level - Who needs to know?'
It shows a small triangle of people, where each descending level involves more people who need to know about the action. The first level is Wholesaler, then Hospital, then Retail, and Consumer is the final layer at the bottom, with the most people who need to know.
You may suggest a market action Class, Type and Level in your notification. These are subject to change during our review in Step 3.
Determining the class of market action
Class I - Critical safety-related
- The problem presents a reasonable possibility of serious injury or death.
- Class I actions must feature the word ‘Critical’ in the title of the customer letter.
Class II - Urgent safety-related
- The problem presents a reasonable possibility of temporary or minor injury, and/or the likelihood of serious injury or death is remote.
- Class II actions must feature the word ‘Urgent’ in the title of the customer letter.
Class III - Lowest risk
The problem is not likely to lead to injury, and/or the chance of a temporary or minor injury is remote. Class III actions usually address non-safety related problems.
How we determine the class
The class of a market action considers the severity of the potential harm, weighed against the likelihood of that harm occurring.
For instance, some harms might be very severe (i.e. a permanent injury) but extremely unlikely, while others might be less severe (i.e. short delay to treatment) but more likely to occur. We consider all scenarios, in particular, the worst-case scenario, when evaluating the potential risk and determining the Class.
Our classification of your action is informed by your submitted risk assessment. You may use the table below to inform your suggested Class, but it should not replace your normal risk assessment approach.
| Likely | Has occurred, is occurring frequently, or is expected to occur again within a short period of time |
|---|---|
| Sometimes | Is expected to occur or reoccur at some time, but not within a short period |
| Rarely | May occur at some point, or a low number of instances expected for a high use product |
| Unlikely | Has not occurred, or an extremely limited number of instances. Very unlikely the harm may ever occur |
| Critical | Very severe injury, likely permanent damage, may require major surgery, potential to be life-threatening if medical intervention is not obtained, or death |
|---|---|
| Serious | Results in more significant injury, impairment requiring professional medical treatment, or the potential of significant sequelae |
| Minor | May result in minor temporary injury or impairment not requiring professional medical intervention |
| Negligible | No risk to health, or extremely mild such as user inconvenience, temporary discomfort with no lasting effects |

{kind=link}
This image shows a risk assessment matrix that uses severity and likelihood to determine risk classifications. The matrix is organised as a table with four rows and five columns.
The vertical axis represents likelihood levels (from top to bottom): Likely, Sometimes, Rarely, and Unlikely. The horizontal axis shows severity levels (from left to right): Negligible, Minor, Serious, and Critical.
The matrix uses a traffic light colour scheme and classifies risks into three classes:
- Class III (shown in green) appears in all Negligible severity scenarios, and for Minor severity when likelihood is Unlikely
- Class II (shown in yellow) covers most Minor and Serious severity scenarios
- Class I (shown in red) represents the highest risk, appearing in all Critical severity scenarios and Serious severity when likelihood is Likely
The classifications follow these patterns:
- Any Critical severity results in Class I, except when Unlikely (Class II)
- Serious severity results in Class I when Likely, and Class II for all other likelihoods
- Minor severity results in Class II, except when Unlikely (Class III)
- Negligible severity always results in Class III regardless of likelihood
This risk matrix helps organisations categorise and prioritise risks based on their potential impact and probability of occurrence.
Deciding the type of market action
Use the information you have gathered in Step 1 to determine the action which would mitigate the problem most effectively.
The types of market action can be understood by asking:
‘What will the customer need to do to address an actual or potential problem with safety, quality, efficacy, performance, presentation or use?’
Recall
‘… return/dispose of the product.’
Recalls permanently remove products from the market.
They include:
- Removing products with design, manufacturing, or safety defects
- Requesting customers to check and return any defective products.
Customers may need to:
- Return products to you or to the place of purchase
- Dispose of the product and contact you for a refund, replacement or credit.
Recalls do not include:
- Removing expired products that were released before expiry
- Removal of an appropriate but small number of products from the market to test for deficiencies.
Product Correction
‘… correct/fix the product.’
Product Corrections address specific or potential deficiencies.
They include:
- Repair, modification, adjustment, or re-labelling of products due to quality, safety, efficacy, performance, or presentation problems
- Updates to accessories, software, operating instructions, patient information leaflets, implant cards, service manuals, and preventative maintenance procedures (even if performed by the customer)
- Corrections to expiry dates, batch numbers, etc.
Some products can remain in use if there is an effective safeguard or workaround until a permanent fix is performed.
The correction can be performed at the customer’s premises or any other agreed location.
Product Corrections do not include:
- Repairs due to incidental malfunction from normal wear and tear or poor customer maintenance
- Preventative maintenance
- Modifications for technical improvements unrelated to quality, safety, efficacy, performance, or presentation.
Product Alert
‘… know something about the product.’
Product alerts raise awareness about deficiencies, potential deficiencies, or other concerns with the use of a product.
They are appropriate when:
- stopping treatment could pose a greater risk than continued use of the affected product i.e. the product provides critical treatment and there are no alternatives.
- the affected product cannot physically be recalled (i.e. implanted therapeutic goods)
- concerns or post-market signals indicate that users are not following established precautions or instructions.
The alert may:
- advise on patient management strategies
- describe precautionary actions for clinicians or users
- detail when alternative products or treatments are expected to be available following investigation
- reiterate existing product safety information for an otherwise safe product
- detail potential/emerging problems with the product.
Product Alerts may later develop into a Recall or Product Correction if alternative products or corrections become available, or if further investigation alters the risk.
Quarantine
‘… avoid use or supply of the product, pending further advice.’
Quarantines temporarily suspend use and/or distribution of products already in the market, pending further investigation of a problem.
It should be considered if a potential defect could affect safety, quality, efficacy or performance of a product.
After issuing the quarantine, your investigation should determine:
- how the problem is occurring
- the risk posed by the problem vs the benefit of continued use
- the proposed next steps.
Following your investigation, we will work with you to determine whether the quarantine can be lifted, or if further action (such as a recall or product correction) is required to address the problem.
If we decide your quarantine can be lifted, we must approve your end-of-quarantine communication before you can distribute it to your customers.
Deciding the level of market action
The level describes who will be notified of the action and usually matches the depth of distribution of the affected product in Australia.
We may adjust the level of the action based on the risk of the problem, or other factors such as avoiding a shortage.
Considerations
- Distribution channels
- How far into the supply chain the affected product is distributed
- Ease of identifying end users
- Whether the action will cause a medicine shortage or medical device supply disruption.
| Wholesale | Hospital | Retail | Consumer |
|---|---|---|---|
| Wholesale level, as well as:
| Hospital and Wholesale levels, as well as:
| Retail, Hospital and Wholesale levels, as well as:
|
Step 3: TGA Review
Our assessment
Our assessment starts when you submit your notification and draft documents. We will send you a TGA agreement letter once we have reviewed and approved your proposed action.
Do not begin contacting your customers until we have sent you the TGA agreement letter and you have returned a signed copy of the TGA-approved version of your customer letter.
Our assessment timeframes
We aim to agree to notifications within 7 business days (not counting the day you notify us). We typically agree within a shorter timeframe, but incomplete, unclear, or unsatisfactory notifications or supporting documents may delay our review and approval.
What our assessment involves
We will objectively review your notification to ensure the proposed action and timeline will effectively mitigate the risk/s. We will consider other factors such as the effect on supply for critical goods.
When we send you our agreement letter, the customer letter/notices we return to you are considered the TGA-approved versions.
We will edit your draft letter if you do not include the following from the TGA customer letter template:
- Correct Title: Include Critical (Class I) or Urgent (Class II) before the action type.
- Clarity: Use plain English, tailored to your audience. Include diagrams or pictures as needed
- Content:
- Do: Present the most important details first, so customers immediately understand what’s the problem and what they need to do.
- Do not: Downplay the seriousness of the risk, promote your company, the affected product/s or other products, or include unnecessary information
- Contact Details: Clearly state how customers can contact you for a refund or arrange replacements/corrections. See Staying Available in Step 4.
If we change the Type or Level of market action, we may inform you prior to sending the agreement letter, because it will impact how you conduct your action.
We may change the Class or formatting of your customer letter/public notice without prior consultation.
If you do not agree with our changes, contact us immediately to discuss.
If we cannot reach agreement on a strategy that would best protect the health and safety of the Australian community, we may use our powers under the Act to mandate a market action.
‘Early Advice’ (communicating with third parties prior to agreement)
If we suspect an action could significantly impact the Australian community or healthcare sector, it may merit ‘an ‘Early Advice Notice’ being sent to the State and territory recall coordinators, relevant health professional guilds and/or peak consumer bodies.
These may:
- raise awareness about upcoming actions to ensure affected stakeholders are aware of mitigation strategies prior to initiation
- seek advice about the appropriateness of the proposed action, including whether it would result in a disruption to patient care or a critical shortage of therapeutic goods.
We advise all recipients that the information:
- is subject to change
- relates to an action that has not yet been agreed or commenced and
- should be provided only to persons who can provide the requested input or advice.
You will be able to review the Early Advice Notice for factual accuracy prior to its distribution.
The length of the Early Advice period may vary based on risk.
TGA web statements
If we determine that your action requires a TGA web statement (see step 4 below), we will draft it and publish it on our website. You will be able to check it for factual accuracy prior to publication.
The TGA agreement letter
The agreement letter summarises the Class, Type, and Level of the market action, and summarises what customers should do.
The agreement letter represents the TGA’s understanding of the action you are going to take and details your reporting timelines and other requirements for the action.
If you do not perform the agreed action as specified, we will follow up with you. The market action is not finished until we accept the conclusions of your closeout report. Follow up reporting is detailed in Step 5.
Step 4: Initiating your market action
When to initiate your market action
When you receive our agreement letter, you should accept all edits to the approved customer letter, sign it, and return it to us before 11am AEST / AEDT the following business day. By doing so, you acknowledge that you have read and understood the contents of the agreement letter and will perform the actions as described therein.
At this point, you may begin distributing the TGA-approved, signed copy of your customer letter to all customers who have received the affected goods.
Your obligations
Distributing other communications
If required:
- Public notices (e.g. sponsor website notices, retail notices, social media posts and/or print advertisements) must be approved by us before you distribute, post, or publish them
- You must notify the ACCC of your action within 2 days of receiving the agreement letter if your therapeutic good is also a consumer good (see section 'Your stakeholders - roles and responsibilities' below).
Staying available
The contact details you provide to customers must include an Australian contact number and/or email address and be responsive to incoming calls / enquiries throughout business hours and public holidays. Staff responsible for monitoring these phones and email inboxes must record outcomes or complaints and have the authority to respond effectively to customers.
Collecting customer responses
Once you have distributed the letter, you should receive confirmation from your customers that they have received, read and understood your customer letter by returning a completed response form.
You must keep records of your customers’ responses, so that you can keep us informed of the progress of your action. This is essential to closing out the action (Step 5).
We prefer that you collect customer responses via post, email, or electronic form.
If you receive acknowledgements via telephone or through a site visit, ensure that you record the name and contact details of the person responding to the action and the date this occurred. We may ask you for these details.
Following up with customers who do not respond
We expect you to attempt at least 3 times (not including your original attempt to contact them) to follow up with customers who do not respond to your customer letter. Use multiple contact methods including email, phone, registered post, and site visits as appropriate.
If a customer is responsive but routinely difficult to reach, consider asking them to review the phone number you have on file for them, and/or whether they could establish a group email inbox, so that your correspondence will be visible to their colleagues.
What we disclose
Notifying states and territories and other stakeholders
Two business days after we send you the agreement letter, we send summary details of the action to the state and territory recall coordinators. Relevant professional guilds and other key stakeholders may also be notified on a case-by-case basis.
This release of market action information ensures that all customers are contacted and this is permitted under our legislation.
Your signed customer letter will also be distributed to the state and territory recall coordinators along with the TGA notification and your customer information.
Publication to the Database of Recalls, Product Alerts and Product Corrections (DRAC)
Two business days after agreeing to a market action, we automatically publish summary details of the action (excluding quarantines, actions involving blood components, or products supplied exclusively for clinical trials) in the publicly searchable Database of Recalls, Product Alerts and Product Corrections (DRAC).
TGA web statements
We also publish information about ‘high profile’ market actions on the TGA alerts page as a web statement. This may be necessary for the following types of actions:
- Consumer level actions
- Product alerts for implanted therapeutic goods
- Actions with broad implications for public health and safety.
Web statements inform and advise consumers and health professionals - in plain language - of appropriate actions to take. We publish them on the TGA website following our agreement to the action with you, typically at the same time as the publication of the summary information about the action to the DRAC.
You will be informed during our assessment if this will happen. The text of the web statement is drafted entirely by us. You will be given the opportunity to review the final draft to confirm that the content is factual.
Statements on your website/social media
If you have been advised to also announce the action on your website, this statement should reflect the wording of the TGA web statement.
Information published on your own website or social media should be publicly available for the length of time specified in our agreement letter (minimum of 3 months). We may adjust this requirement when we review your closeout report.
Step 5: Finalising your market action
Reporting requirements
You are required to periodically report to us about the progress of your action. Additional reporting requirements may be imposed at our discretion.
- You must use the reporting templates included in the agreement letter. They are also available on our website – see the templates page
- These reports are typically due at 6 weeks (interim) and 12 weeks (closeout) after commencing the action
- Different reporting timeframes may be agreed on a case-by-case basis
- If the action is not completed by the 12th week, you will be asked to provide a valid reason for the delay and additional reports at a frequency determined by us
- For certain actions, such as for blood component recalls, we may require only one report. The due date will be provided in the agreement letter
- You may submit the closeout report earlier if all actions are complete (all goods returned/corrected, the root cause investigation and CAPA are finalised).
Your obligations when there has been overseas supply
Within 10 days of agreement, you must notify the Australian Government Minister for consumer safety about the overseas supply.
This requirement is in addition to your obligation to notify the Minister within 2 days of initiating a market action in Australia for a therapeutic good which is also a consumer good. (S128(4); Competition and Consumer Act 2010).
Common mistakes when reporting on market actions
Your follow up reports must be thorough and objective and demonstrate that the market action was effective. We will follow up with you if your reports are inadequate.

{kind=link}
This image presents a guidance table for completing incident reports, organised into two columns labelled "Do" and "Do not" with four main categories:
- Action progress
- Root cause
- Remedial action
- Response rates
The table uses tick marks (✓) to indicate recommended actions and cross marks (✗) for actions to avoid.
Under Action progress:
Do: Provide certificates of destruction or consignment documents where available.
Do not: Omit a certificate of destruction without explaining why it is not available.
Under Root cause, there are 3 "Do" items:
- Outline the root cause of the problem as discovered in your investigation.
- Clearly describe the root cause of the problem in plain English.
- Explain the most likely root cause if you could not find a definite one.
The corresponding Root cause "Do not" items are:
- Reiterate the problem statement instead of describing the root cause.
- Provide inadequate explanation. Answers such as “software defect” or “product contamination” do not provide enough information.
- Cite the TGA as the reason for the action.
Under Remedial action there are 2 "Do" items:
- Explain how the manufacturer's solution will address the root cause described.
- Clearly describe what has changed in the manufacturing process or product’s design to prevent reoccurrence (i.e. CAPA)
The corresponding Remedial action "Do not" items are:
- Provide inadequate explanation. Answers such as “software upgrade” or “new design” do not provide enough information.
- Claim that the root cause has been addressed by an IFU update if the problem is not due to user error.
Under Response rates:
Do: Explain how customers have been contacted, and how many follow up attempts were made with non-responders.
Do not: Report a low number of total responses without explaining efforts to contact end users.
If you repeatedly submit poor quality reports, or do not submit reports at all, we may use our legislative powers to compel you to submit specified information within a set timeframe.
When we receive your closeout report
If we do not believe that you have sufficiently addressed the problem, or adequately attempted to reach non-responders, we will follow up with you for more information.
If/when we accept your closeout report, you will receive an email and final letter confirming we have closed your action on our database. At that point, no further information is required by you.
If you have not been able to recall/correct all products, we may still close the action, as long as you can provide sufficient evidence that you have been thorough in attempting to contact all non-responders.
If a non-responding customer contacts you after we’ve closed the action, you must still recall or perform the corrective actions for that product's service life or until a medicine expires. For consumer goods, you must also comply with the Australian Consumer Law (ACL).
Your stakeholders - roles and responsibilities
The following section details how we expect sponsors to interact with different stakeholders involved in market actions.
You are responsible for initiating and conducting market actions in Australia. You may optionally authorise a third party to perform the action on your behalf.
Your market action procedure
Your Quality Management System (QMS) should outline your market action procedure:
- Responsible person(s) in your organisation
- What they should do in an emergency (noting our immediate actions process - see top of page)
- How they should conform to this procedure
- Where they will keep distribution records (a condition of entry on the Australian Register of Therapeutic Goods)
- Arranging return of goods and/or disposal
- Customer reimbursement policy
- Where you store key information (e.g. contact details for the TGA, state and territory recall coordinators, etc).
Mock recalls
Simulated or mock recalls can help you prepare for a future action, but they are not required to be reported to us. If you wish to submit a simulated recall through the TBS Portal for practice, please clearly mark it as Mock Action for Training Purposes Only.
Your manufacturers
You should have written agreements with your manufacturer(s) to ensure you can both respond to potential market actions. Your manufacturer(s) should be willing to work with you to investigate and address problems with therapeutic goods.
| Medicine manufacturers | Medical device manufacturers | Biological, human blood and blood component manufacturers |
|---|---|---|
Part I of the PIC/S Guide to GMP for Medicinal Products addresses recalls and complaints.
| ISO 14971 Medical devices - application of risk management to medical devices establishes responsibility for analysing potential risks associated with adverse events, goods failure or complaints. ISO 13485 Medical devices - Quality management systems - Requirements for regulatory purposes, also requires systems for:
| Are required by the Code of GMP for human blood and blood components, human tissues and human cellular therapy products to:
|
Risk analyses
Manufacturers are responsible for investigating problems (risk assessments, root cause analyses, etc.) that occur during the manufacturing process. Sponsors can also investigate problems, but you should have a clear agreement in place with your manufacturer if you are going to investigate a problem on their behalf.
If you are also a manufacturer
You should follow the requirements for both sponsors and manufacturers.
Australian manufacturers with a TGA licence
It is a condition of the licence that manufacturers inform us promptly if they intend to initiate a recall.
Your wholesalers and distributors
Your wholesalers and distributors should have procedures to respond effectively to market actions.
Wholesalers of scheduled medicines should follow the Australian Code of Good Wholesaling Practice for Medicines in Schedules 2, 3, 4 and 8.
Distribution records
Your wholesalers and distributors should keep records sufficient to track any batch of goods from the distribution chain.
All distribution records should clearly note the batch and date. They should be provided to you immediately upon request. You should ensure that your wholesalers and distributors are aware of this obligation.
Wholesaler/distributor’s market action procedures
Your wholesaler/distributor should have the following in place in the event of a market action:
- Established responsible contact person(s)
- Processes for:
- Tracing batches within the stock control system
- Quarantining affected goods
- Handling returns, replacements (where appropriate) and credit
- Record keeping (including lists of all customers and their contact details).
Once your action has been initiated, you should instruct your wholesaler/distributor to provide your customer letter to their customers as soon as possible.
You should have an agreement in place with your wholesaler establishing which party is responsible for noting the responses from those customers for your reporting requirements.
State and territory recall coordinators
State and territory recall coordinators maintain their own alert systems and procedures for communicating market action information to their jurisdictions. This system includes contact details for relevant organisations and health professionals who may be affected.
If you cannot reach a customer when attempting to follow up with them, you may contact a state or territory recall coordinator to see if they have more up-to-date contact information.
Australian Competition and Consumer Commission (ACCC)
The ACCC is Australia’s national competition, consumer, fair trading, and product safety regulator. One of their roles is to identify and address the risk of serious injury and death from safety hazards posed by ‘consumer goods’.
The Australian Consumer Law (ACL) defines ‘consumer goods’ as goods intended to be used, or are of a kind likely to be used, for personal, domestic or household use or consumption.
Some therapeutic goods are also consumer goods.
Will the TGA or the ACCC be the lead regulator for my market action?
‘Lead Regulator’ means the regulator responsible for coordinating the action.
For therapeutic goods that are also a consumer goods, the ACCC will be the lead regulator for actions involving non-compliance with a safety standard or ban. The ACL Part 3-3 explains bans, and Part 3-4 explains safety and information standards.
In all other cases, the TGA remains the lead regulator for the problem. If the therapeutic good is also a consumer good, you must notify the ACCC within 2 days of beginning your action.
You may refer to our flowchart on the following page to determine who the lead regulator is for a given problem.
How do I report a problem to the ACCC?
To notify the ACCC of a market action, go to the submission portal on the ACCC website and complete their online form.
The ACCC receives this notice on behalf of the Australian Government Minister responsible for the ACL (refer to Section 128 of the ACL, Schedule 2, Competition and Consumer Act 2010).
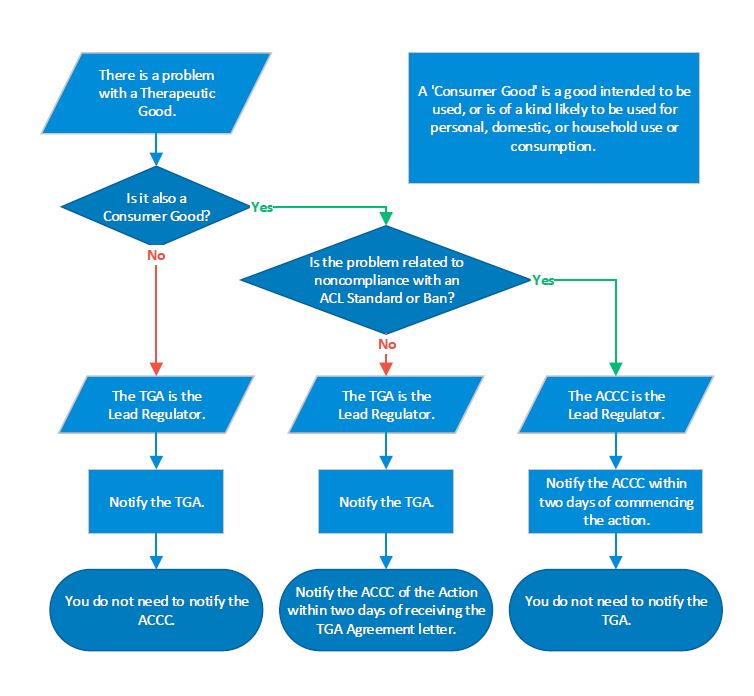
Flowchart and examples
{kind=link}
A flowchart explaining how to determine the lead regulator and who to notify when handling problems involving therapeutic goods that are also consumer goods.
If there is a problem with a therapeutic good that is not a consumer good, the TGA is the lead regulator, and the ACCC does not need to be notified of the action.
If the therapeutic good is also a consumer good, but the problem does not relate to noncompliance with a standard or ban as defined by the ACL, then the TGA is still the lead regulator, but the ACCC should also be notified within two working days of commencing the action.
If the therapeutic good is also a consumer good, and the problem does relate to noncompliance with a standard or ban as defined by the ACL, then the ACCC is the lead regulator, and the TGA does not need to be notified.
Example
Sponsor A discovers a problem with 'Pregnancy Testing Kit A’. The battery compartment is not secure, the sponsor has received multiple complaints about batteries falling out of the device.
'Pregnancy Testing Kit A’ is powered by button batteries. All required warnings are provided on the packaging to indicate that the device contains button batteries.
Before submitting the action to the TGA, the sponsor checks the ACCC’s Product Bans and Mandatory Standards webpage and finds that because the battery compartment is not secure, the product does not comply with the Mandatory Standard for products containing Button Batteries.
Instead of submitting a notification to the TGA, the sponsor notifies the ACCC that it is recalling the product within 2 days of taking the action.
What if the defect was not related to an ACCC product ban or mandatory standard?
If 'Pregnancy Testing Kit A’ had a secure battery compartment but was experiencing a problem unrelated to ACCC product bans or mandatory standards, then the TGA would be the lead regulator for that action.
The sponsor is still required to notify the ACCC within 2 days of taking action, because the kit is defined as a therapeutic good AND a consumer good.
In practice, Sponsor A should notify the ACCC when they receive the agreement letter from the TGA.
What if I report the problem to the wrong regulator?
Notifications incorrectly submitted to us which relate to ACCC Product Bans and Mandatory Standards will be referred to the ACCC, and we will inform you when we do this. Likewise, notifying the ACCC of a problem for which we are the lead regulator will result in the case being referred to us.
To avoid unnecessary delays, familiarise yourself with the definition of ‘consumer goods’ and the following (non-TGA) resources:
Page history
Updates to reflect that the PRAC is now in effect and changes based on stakeholder feedback, including:
- Clinical trials included under ‘Immediate actions’
- Contact details updated to include accessibility options
- New graphics for ‘the end-to-end market action process’ and ‘documents you must include’
- ‘Product identifiers’ updated to include GTIN and CTN/CTA
- Minor updates to the sections ‘Electronic response forms’, ’When to initiate your market action’ and ‘When we receive your closeout report’
- Other minor editorial changes
Original publication as a preview ahead of the PRAC coming into effect.
Updates to reflect that the PRAC is now in effect and changes based on stakeholder feedback, including:
- Clinical trials included under ‘Immediate actions’
- Contact details updated to include accessibility options
- New graphics for ‘the end-to-end market action process’ and ‘documents you must include’
- ‘Product identifiers’ updated to include GTIN and CTN/CTA
- Minor updates to the sections ‘Electronic response forms’, ’When to initiate your market action’ and ‘When we receive your closeout report’
- Other minor editorial changes
Original publication as a preview ahead of the PRAC coming into effect.