Reclassifying surgical mesh devices
Guidance on reclassification of surgical mesh devices in Australia, outlining regulatory changes to Class III, ARTG application requirements, transition arrangements, and sponsor obligations.
Purpose
This guidance is for sponsors of surgical mesh medical devices, and to assist the TGA's assessors when dealing with device applications and entries affected by the re-classification requirements.
If you import and/or supply these devices, you need to know about the regulatory changes that will impact your product.
The Australian Government has strengthened the assessment of surgical mesh medical devices by approving regulatory amendments that reclassified all these medical devices from Class IIb (medium) to Class III (high risk).
This decision is consistent with the recommendations from the Expert Review of Medicines and Medical Devices Regulation (MMDR) to align (wherever possible) the Australian classification of medical devices with the European Union framework.
Introduction
The Australian Government has made the decision to reclassify surgical mesh devices ahead of Europe due to the serious concerns about risks associated with the use of these devices.
We announced the regulatory amendments on 26 October 2017. The Therapeutic Goods (Medical Devices) Amendment (Implantable Medical Devices) Regulations 2017 provide that from 1 December 2018, surgical meshes are classified as Class III medical devices as follows:
Clause 3.4 - Surgically invasive medical devices intended for long-term use and implantable medical devices
(4A) The device is classified as Class III if it is:
(b) surgical mesh
These amendments do not affect any surgical mesh medical devices that contain tissues, cells or substances of animal, or microbial, or recombinant origin, or stable derivatives of human blood or human plasma, or a substance that, if used separately, might be considered to be a medicine, or devices intended to be wholly, or mostly, absorbed by a patient's body or undergo a chemical change in the body, as these devices are already Class III medical devices in accordance with Therapeutic Goods (Medical Devices) Regulations 2002.
Surgical mesh medical devices
Surgical mesh medical devices are a variety of surgical implants usually used in the repair of soft tissue, or mixed soft tissue/bony structural defects, to support organs and/or tissues or to strengthen the integrity of a body cavity.
Surgical mesh is implanted in the body for repair or reinforcement and includes tapes and slings.
Surgical mesh devices can be used in a range of anatomical locations and structures.
These include, but are not limited to, repair and/or support to tissue and/or organs of the perineum, urethral structures, pelvis, abdomen, breast, and soft tissue of the inguinal, femoral, and ventral regions.
Surgical mesh devices are also used in ligaments, tendons, and other periarticular tissue in repair and/or support.
They may be constructed from a range of absorbable or non-absorbable materials (e.g. metallic mesh, flexible synthetic or organic materials etc.), may have different forms (e.g. solid sheet, matrix, woven or knitted, etc.) and shapes (e.g. sheet, sling, tape, etc.).
Surgical meshes are generally designed to be integrated into the structures they support.
The primary mechanical function can be either unidirectional (longitudinal) tensile strength or multi-directional support or strength.
Examples of surgical mesh medical devices
Hernia surgical mesh
Types of Hernias:
- inguinal - occurs in the inner groin
- femoral - occurs in the upper thigh/outer groin
- incisional - occurs through an incision or scar in the abdomen
- ventral - occurs in the general abdominal/ventral wall
- umbilical - occurs at the belly button
- hiatal - occurs inside the abdomen, where the stomach pushes up through the diaphragm.
Urogynaecological surgical mesh
Most commonly indicated for pelvic organ prolapse and stress urinary incontinence.
Male sling
Stress urinary incontinence
Surgical mesh used in orthopaedic and trauma reconstructive surgeries
For example, synthetic mesh for tendon repair, tissue approximation during reconstruction of damaged or torn ligaments, tendons or other soft tissues.
Synthetic or biological surgical mesh patches used in plastic and reconstructive surgery
For example, mesh intended acting as a barrier hindering the migration of the cells and tissues, support devices in breast surgery.
- Abdominal plugs made from mesh.
Mesh-like implants used for compensation of bone loss or repair of structural bone defects (e.g. acetabulum cages, cranial mesh, or mesh used in facial (mandibular or zygomatic) bones repair) are not considered as surgical meshes.
Timing
Transition arrangement changes
On 29 October 2021, amendments to the Therapeutic Goods (Medical Devices) Regulations 2002 came into effect that removes the requirement for notification to the TGA about surgical mesh devices that are subject to the reclassification.
In addition, for surgical mesh devices (other than urogynaecological surgical mesh devices), an application for TGA conformity assessment will be sufficient to enable the continued supply of surgical mesh devices under current Class IIb ARTG entries.
Actions to consider
| Date | ARTG inclusion applications submitted on or after 1 December 2018 | Pre-commencement entries |
|---|---|---|
| Before 1 December 2018 | N/A | Review all your existing ARTG entries for surgical mesh medical devices and/or all applications you have submitted to the TGA seeking pre-market authorisation for your surgical mesh medical devices. |
| On or after 1 December 2018 | All applications will be for Class III ARTG inclusion only | |
| By 1 December 2020 | N/A
| If you have a urogynaecological mesh entry in the ARTG and you want to continue supply the device, you must submit an application for inclusion of your medical device in ARTG as a Class III by this date to the TGA. |
| By 1 December 2021 | If your medical device is any other type of surgical mesh, you must have an appropriate TGA conformity assessment certificate issued after 1 June 2021 or submit an application for appropriate TGA conformity assessment by this date; or have an application for inclusion in the ARTG as a Class III by this date. Note this is required irrespective of the existing risk profile of the device. If the TGA did not receive your application for appropriate conformity assessment or Class III application for inclusion in the ARTG within this prescribed period, the transitional provision will no longer apply to your device. |
New medical device applications
Any application for inclusion of a surgical mesh in the Australian Register of Therapeutic Goods (ARTG), submitted to the TGA on or after 1 December 2018, must be submitted as an application for a Class III medical device.
You must use the Class III TGA Business Services Device Application form.
Any application for ARTG inclusion using a form other than a Class III TGA Business Services Device Application will not pass preliminary assessment1.
Transitional provisions for pre-commencement entries
Devices to which transitional provision will apply
Sponsors will be able to access the transitional provision only if the following requirements are met:
- Only sponsors who have pre-commencement entries are able to access the transitional provisions. A pre-commencement entry is an ARTG inclusion of any 'kind of medical device' that is both:
- included in the ARTG as a Class IIb medical device following assessment of an application made before 1 December 2018, and
- regardless of whether the commencement date for the ARTG entry was before or after 1 December 2018.
- The application for appropriate TGA conformity assessment certificate for the individual devices covered under the Class IIb entry has been submitted between 1 July 2020 and before 1 December 2021, or
- The application for appropriate TGA conformity assessment submitted between 1 July 2020 and 1 December 2021, is not withdrawn before 1 December 2021, has not lapsed, and if the certificate was issued before 1 December 2021 it was within 6 months of 1 December 2021 and
- An application for inclusion as Class III medical device is submitted within 6 months after the TGA conformity assessment certificate is issued.
- An application for inclusion has not been made for the kind of medical device that is currently under assessment in the application for appropriate TGA conformity assessment.
IMPORTANT
If you do not comply with these requirements by the due date as applicable, your devices will no longer be eligible for the transitional provision, and you will be required to stop importation and/or supply of your devices immediately.
If you decide to seek pre-market authorisation for your device at a later date, you will be required to submit an application for inclusion of a Class III medical device in ARTG as per the usual application process.
If you are unsure whether your device is a surgical mesh medical device, and therefore whether you are required to reclassify your device, we advise that you provide information about your device to the TGA and give reasons on why you consider your device is not subject to the reclassification requirements.
Application for inclusion of the surgical mesh in ARTG as Class III
After you obtain a valid conformity assessment certificate, you will be required to submit applications for inclusion of your surgical mesh devices in the ARTG as Class III medical devices prior to the end of the transition period:
- For urogynaecological mesh: by 1 December 2020
- For all other surgical mesh: After the earlier of either:
- The day the TGA conformity assessment application is withdrawn
- The day the application for appropriate TGA conformity assessment application lapses
- The day a decision to refuse issuing a TGA conformity assessment certificate for the kind of medical device in the application becomes final and is no longer subject to appeal or
- Within six months of the conformity assessment certificate being issued.
Examples of how the transitional provision will apply
Example A: Other surgical mesh device
- ARTG entry: Class IIb
- GMDN: Male stress urinary incontinence surgical mesh
- ARTG Start date: 22 October 2017
Sponsor has 3 devices of the kind included in this ARTG entry with 3 different UPIs.
- Submit an application for appropriate TGA conformity assessment certificate for all 3 devices of the kind you have in your Class IIb ARTG entry before 1 December 2021, or
- Submit three Class III applications for inclusion of each individual surgical mesh (individual UPI) in ARTG by no later than 1 December 2021 or up to 6 months after the date of issue of a TGA conformity assessment certificate for the surgical mesh.
If you meet either requirement within the specified timeframes, you may continue supplying your devices in Australia until all your respective Class III applications are finalised.
If your Class III applications are successful, your respective surgical mesh devices will be re-included in the ARTG as three individual Class III medical device entries.
After you receive notifications of your new Class III ARTG entries, you may request TGA to cancel your related Class IIb ARTG entry. Otherwise, TGA will make a decision regarding cancellation of your Class IIb ARTG entry on our own initiative.
If one (or all) of your Class III applications for inclusion in the ARTG or for TGA conformity assessment are unsuccessful (lapsed, withdrawn, or a decision is made not to include the device in ARTG, or the TGA refuses to issue a conformity assessment certificate for the device), or after the TGA conformity assessment certificate is issued, you will no longer be able to supply the device(s) in Australia.
The TGA will make a decision regarding the cancellation of your respective Class IIb ARTG entry.
Example B: Other surgical mesh device
A Class IIb ARTG inclusion application (GMDN: Abdominal hernia surgical mesh, synthetic polymer) is submitted to the TGA on 1 November 2018;
and
The application is finalised, and the kind of device is included in the ARTG on 1 April 2019.
After you have received your ARTG entry, you may start importing and supplying your medical device(s).
However, in order to continue to be eligible for the transitional provision, you must:
- Submit an application for appropriate TGA conformity assessment certificate before 1 December 2021, or
- Submit a Class III application for inclusion of each individual surgical mesh (individual UPI) in the ARTG before 1 December 2021 or no later than six months after the date of issue of conformity assessment certificate.
For the remaining part of the process, refer to Example A.
If at least one of the requirements (notifying the TGA and/or submitting a Class III ARTG inclusion application) is not met by the relevant date as specified in Therapeutic Goods (Medical Devices) Regulations 2002, you will no longer be able to manufacture, import and/or supply your surgical mesh in Australia until you reapply and (if successful) re-include your device in ARTG as a Class III.
Kind of device
Only devices of the same kind can be supplied under one ARTG entry.
Class IIb medical devices
Class IIb devices are taken to be of the same kind as another medical device if these devices:
- are supplied by the same sponsor
- are manufactured by the same manufacturer
- have the same medical device classification
- have the same Global Medical Device Nomenclature (GMDN) System Code.
Class III medical devices
In addition to the above characteristics, devices of the same kind must have the same unique product identifier (UPI).
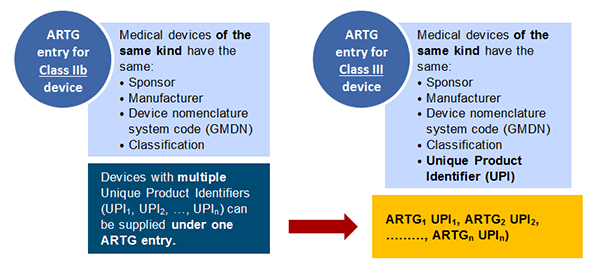
{kind=link}
A diagram comparing Australian Register of Therapeutic Goods (ARTG) entry requirements for Class IIb and Class III medical devices.
On the left, a blue circle labeled "ARTG entry for Class IIb device" lists four common elements for medical devices of the same kind:
- sponsor
- manufacturer
- device nomenclature system code (GMDN)
- classification
On the right, a blue circle labelled "ARTG entry for Class III device" includes the same four elements as Class IIb, plus an additional item:
- Unique Product Identifier (UPI)
Below the circles, a note explains that devices with multiple Unique Product Identifiers (UPI1, UPI2, ..., UPIn) can be supplied under one ARTG entry.
An arrow points to a yellow box at the bottom, illustrating how multiple devices (ARTG1 UPI1, ARTG2 UPI2, through to ARTGn UPIn) can be listed under a single entry.
This diagram highlights the key difference between Class IIb and Class III devices in the ARTG system, with Class III requiring a Unique Product Identifier.
Note
The devices of the kind you have included in your Class IIb ARTG entry may have different UPIs, and therefore may no longer be of the same kind as Class III medical devices.Each Class III medical device with individual UPIs will require a separate application for inclusion in ARTG.
A Class III ARTG entry may still cover more than one device, but only if the:
- devices have the same UPI
- variation in the design of the devices is only to accommodate different patient anatomical requirements
- variation does not change the intended purpose of the device (for example, surgical meshes of the different size).
Preliminary assessment - Requirements for Class III applications for ARTG inclusion
For a Class III medical device application to pass preliminary assessment:
- the application must be made electronically via the TGA Business Services (TBS) client's portal
- it must be made in accordance with a form and manner approved for the classification of a medical device (there are separate forms approved for different classifications and as the sponsor, you must select correct form for the Class III device). For further information, go to Application requirements for medical devices - preliminary assessment
- the application fee must be paid
- the application must be accompanied by the kind of information prescribed for the application of this classification by legislative instrument and provided in an approved form
- you must have the appropriate conformity assessment documents demonstrating the application of the conformity assessment procedures or comparable procedures to the device and provide these documents with the application for inclusion of your medical device in ARTG. For further information, go to Application requirements for medical devices - preliminary assessment
- for devices that must have a TGA conformity assessment certificate - such certificate is in force
- the applicant must certify the section 41FD matters.
The Class III application fee is payable.
Application fees are listed in the TGA's Schedule of fees and charges.
Conformity assessment documents
For the purposes of applications for ARTG inclusion of Class III medical devices, the TGA will consider documents issued by:
- Notified Bodies designated under European Union medical device regulatory frameworks
- United States Food and Drug Administration (FDA)
- Health Canada
- Relevant Japanese authorities (the Ministry of Health, Labour and Welfare (MHLW), Pharmaceutical and Medical Devices Agency (PMDA) or Registered Certified Body (RCB), whatever is applicable)
- Medical Device Single Audit Program (MDSAP) Auditing Organizations (AO)
The documentation must be issued for the same medical device as the one you are applying for in Australia with:
- the same design
- the same intended purpose, and
- intended for the same clinical indications.
The conformity assessment procedures, or comparable procedures, set out the requirements relating to the application of manufacturers' quality management systems for medical devices and other requirements imposed on manufacturers, specifically in relation to the design and production of medical devices.
Conformity assessment procedures that must be applied to a Class III medical device intended by the manufacturer to be supplied sterile are either:
- the full quality assurance procedures (including design-examination) or
- the type examination procedures and the production quality assurance procedures (Regulation 3.6(2) of the Therapeutic Goods (Medical Devices) Regulations 2002).
Respectively, examples of conformity assessment documents that are prescribed for Class III applications for inclusion of medical devices in the ARTG are any of the following2:
- TGA conformity assessment certificate issued either under Part 1 (including Clause 1.6), or under Part 4 and Part 2 of Schedule 3, Therapeutic Goods (Medical Devices) Regulations 2002
- EC Certificate issued by notified body within the meaning of Council Directive 93/42/EEC either under Annex II (including section 4), or under Annex V and Annex III
- EU quality management system certificate issued either under Chapter I of Annex IX and EU technical documentation assessment certificate issued under Chapter II of Annex IX, or production quality assurance certificate issued under Part A of Annex XI and EU type-examination certificate issued under Annex X, issued by a notified body within the meaning of the EU medical devices regulation
- MDSAP certificate or a quality management system certificate for the purposes of the Japanese PMD Act, and a pre-market approval issued under the Japanese PMD Act
- MDSAP certificate and Class IV medical device licence issued under the Canadian medical devices regulations
- MDSAP certificate and an order approving an application for premarket approval under section 515 of the US FDC Act (PMA).
If you require clarification about conformity assessment documents that you need to provide with your application for inclusion of your surgical mesh medical device in ARTG as a Class III, or if you are not sure if the conformity assessment documents that the manufacturer has are appropriate, you may contact the TGA and seek advice.
TGA will consider your manufacturer's conformity assessment documents and will discuss possible options with you.
Providing correct conformity assessment documents is one of the important requirements to ensure your application is not refused.
Audit assessment of Class III applications
Applications for ARTG inclusion of Class III medical devices (for which a TGA-issued conformity assessment certificate is not in force) must be selected for audit.
The level of audit (Level 1 or Level 2) may depend on the category of a medical device and the overseas regulator conformity assessment document(s) provided with the application.
Class III applications must be selected for audit, and the audit assessment fee is payable.
When your application is selected for audit, you must provide all the necessary information to demonstrate compliance with the Essential Principles to the TGA for assessment when requested.
At minimum, you will be required to provide:
- labelling
- instructions for use
- manual and product brochure, and
- clinical evidence for the device.
Sponsors can submit requests with their applications requesting abridgement of the audit assessment (including requests to abridge the level of audit if appropriate):
- sponsors are required to provide justification supporting their requests, and
- TGA's delegate will decide whether such request is appropriate.
Guidance on requests for abridged assessment is available.
Example of 'level of audit' decision
An ARTG inclusion application is submitted for Class III medical device for which the manufacturer has US FDA Pre-Market Approval (PMA) (there is no TGA-issued conformity assessment certificate for this device).
The Class III ARTG inclusion application passes preliminary assessment and it must be selected for application audit.
Level of application audit
A decision on the audit level is based on information provided in the application and/or information or signals that the TGA has regarding the device (or similar devices):
The application will be selected for Level 2 audit if:
- the device does not appear to be the same as the device specified in the PMA (including differences in the UPI, design, intended purpose or clinical indications)
- there are other signals that raise safety or performance concerns.
It is your responsibility to demonstrate that the overseas conformity assessment document has been issued for exactly the same medical device.
The application will be selected for Level 1 audit if none of the criteria for Level 2 audits apply.
You must comply with the Essential Principles and must have sufficient information to demonstrate compliance.
Fees and charges associated with reclassification
The Class III application fee and compulsory audit assessment fee will be applicable to all Class III applications for surgical mesh medical devices.
If the device is included on the ARTG, Class III annual charges will apply.
See Summary of fees and charges to applications submitted to the TGA.
Device reclassification - amendments to Prostheses List
Some sponsors list their devices on the Prostheses List in order for the devices to be reimbursed by private health insurers.
When we reclassify a medical device, a new ARTG entry is assigned.
If your Class III ARTG inclusion application is successful and you received a new ARTG entry, you will be required to update your Prostheses List listings in order to remain on the Prostheses List.
Failure to maintain the currency of 'Billing code' details in the Prostheses List may result in discrepancies between private hospital records and the Prostheses List.
These discrepancies may result in benefits not being paid by private health insurers.
To update the Prostheses List with your new ARTG numbers, you will need to submit an Amendment Application using the Health Products Portal (HPP) for each of the affected billing codes.
When submitting an amendment application, ensure that you clearly advise the reason for the change and include relevant supporting documents that clearly identify the required changes.
Examples of supporting documentation include:
- new ARTG certificate
- catalogues with any changes highlighted.
For any queries about amending your Billing code on the Prostheses List, contact the Prostheses Hotline by phone on (02) 6289 9463 or by email at prostheses@health.gov.au.
Frequently asked questions
What will happen after successful completion of the assessment of my Class III application?
If your application(s) for reclassification of surgical mesh devices are successful, and all your devices are included in ARTG as Class III, you will no longer require a Class IIb ARTG entry, and this entry may be cancelled.
We encourage you to request cancellation as soon as possible to prevent paying unnecessary annual charges. Otherwise, the TGA may make a decision regarding cancellation on our own initiative.
You will continue to be required to pay annual charges for the Class IIb ARTG entry until it is cancelled.
What will happen if my Class III application is not successful?
If your Class III application is not successful, you will be notified of the decision in writing and you will be provided the reasons for the decision.
If you are not satisfied with this decision, you may request reconsideration of this initial decision under section 60 of the Therapeutic Goods Act 1989 within 90 days of the decision.
If you are not satisfied with the reconsideration (reviewable decision), you may apply to the Administrative Appeals Tribunal or the court.
Once the final decision regarding your Class III application is made3, the TGA will make a decision regarding cancellation of your related Class IIb ARTG entry.
Can I continue supply of my surgical meshes in Australia after the end of the transitional period if the audit assessment of my Class III application is not finalised?
You can continue supply of your devices under the Class IIb ARTG entry until the decision on your Class III application(s) is made, or if the application is withdrawn or has lapsed, if you:
- have submitted an application for appropriate TGA conformity assessment certificate before 1 December 2021
- submitted Class III ARTG inclusion application(s) before 1 December 2021, or within six months of the date of issue of the TGA conformity assessment certificate for the kind of device
- your Class III application for inclusion in the ARTG or appropriate application for TGA conformity assessment certificate for the individual devices covered under the Class IIb entry submitted between 1 July 2020 and before 1 December 2021 is still under assessment by the TGA.
Footnotes
- To pass preliminary assessment, an application must meet the requirements in section 41FDB of the Therapeutic Goods Act 1989. The TGA will carry out an assessment of whether the requirements have been met for each application. Applications that do not pass preliminary assessment will be refused.
- For further references on the information that must accompany the application for ARTG inclusion, refer: Therapeutic Goods (Medical Devices - Information that Must Accompany Application for Inclusion) Determination 2018 and the respective guidance document 'Use of market authorisation evidence from comparable overseas regulators/assessment bodies for medical devices'.
- Finally determined means that the decision has been made and there is no longer any possibility of a change in the outcome of that decision, refer to subregulation 11.29(5) of the Therapeutic Goods (Medical Devices) Regulations 2002.
Page history
Title changed from 'Reclassification of surgical mesh devices' to 'Reclassifying surgical mesh devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Amendment to transition arrangements due to amendment to Therapeutic Goods (Medical Devices) Regulations 2002.
Clarifying the meaning of surgical mesh and giving more examples of what is and isn't surgical mesh. Minor editorial changes.
Title changed from 'Reclassification of surgical mesh devices' to 'Reclassifying surgical mesh devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Amendment to transition arrangements due to amendment to Therapeutic Goods (Medical Devices) Regulations 2002.
Clarifying the meaning of surgical mesh and giving more examples of what is and isn't surgical mesh. Minor editorial changes.