We’re experiencing issues with our datasets and search. We apologise for any inconvenience while we work to fix this.
The new Procedure for Recalls, Product Alerts and Product Corrections (PRAC), took effect on 5 March 2025.
The PRAC replaced the Uniform Recall Procedure for Therapeutic Goods (URPTG) as the procedure for sponsors to follow when conducting market actions in Australia.
What’s changed from the URPTG to the PRAC?
While the PRAC is a complete re-write of the URPTG to improve its content and readability, the process for sponsors performing a market action largely remains the same.
Some of the changes introduced in the PRAC include:
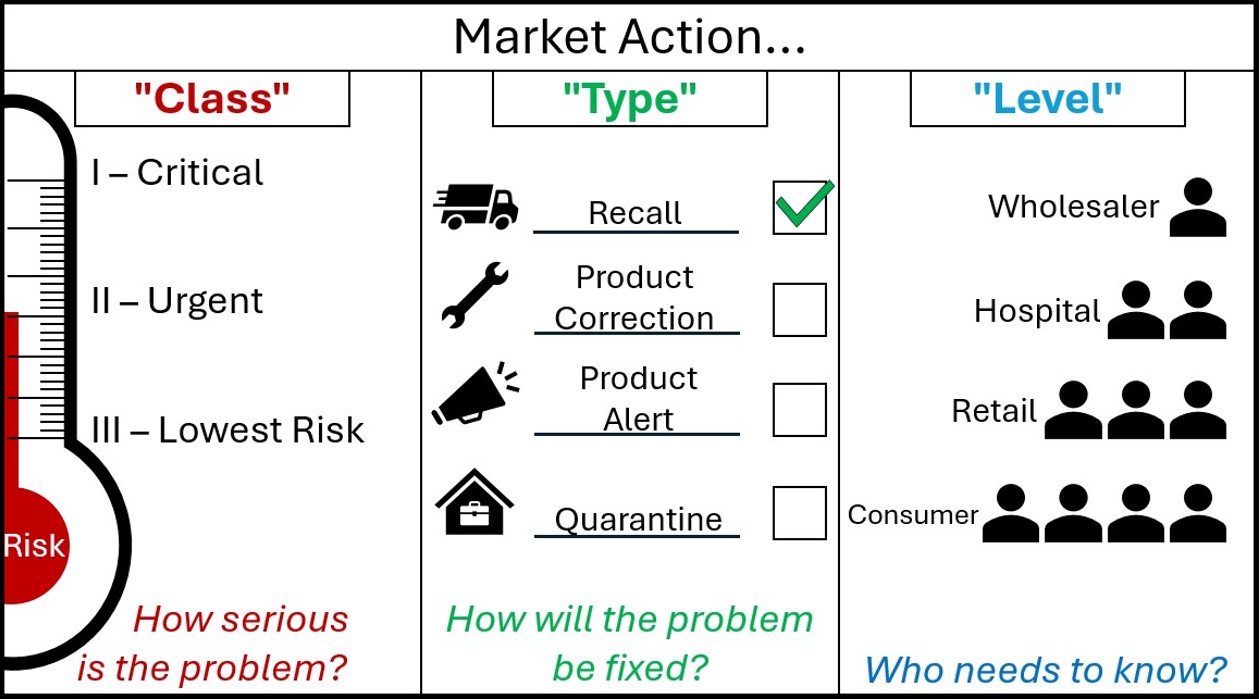
- updating the recall terminology, removing the sometimes-confusing categories of ‘recall’ and ‘non-recall’ actions, and replacing them with one category called ‘market actions’
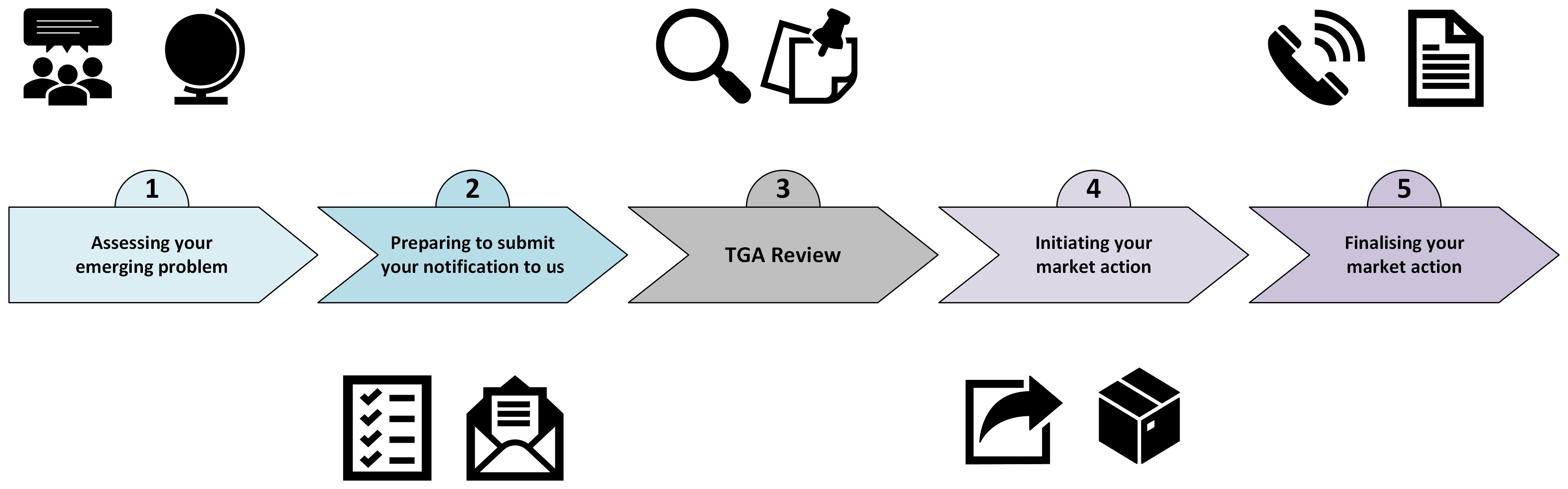
- condensing the document and reducing the number of steps in the recall process from 10 to 5
- placing information in easy to digest formats including tables or pictures, where appropriate
- clarifying when we require extra information for actions which may create a shortage, or involve lengthy corrections, etc
- simplifying the definitions for Class and Level of market actions without changing their meanings
- removing information concerning our legislative powers related to recalls. This information will be updated and placed as a separate guidance piece on the TGA website when the PRAC takes effect
- new processes where we will:
- distribute sponsors’ approved customer letters with our TGA notification to the state and territory recall coordinators
- no longer verify the accuracy of sponsor customer lists through protracted back-and-forth dialogues. Sponsors are expected to submit accurate information to avoid delays.
The PRAC retains the new reforms introduced in the final version of the URPTG (Version 2.4) in March 2024, including flexible reporting requirements, greater transparency around the 'Early Advice' process, and new user-friendly templates.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}