Understanding regulatory requirements for in vitro diagnostic (IVD) companion diagnostics (CDx)
Guidance on the regulatory framework for in vitro diagnostic (IVD) companion diagnostics (CDx) and for the medicine or biological that requires CDx testing.
Purpose
The purpose of this guidance is to outline the regulatory framework for in vitro diagnostic (IVD) companion diagnostics (CDx) and for the medicine or biological that requires CDx testing.
It provides information on clinical and analytical performance requirements for IVD CDx.
The guidance is applicable to both medical device and medicine sponsors.
This guidance addresses requirements for sponsors of commercial IVD CDx and for pathology laboratories with in-house IVD CDx.
However, this guidance does not detail laboratory accreditation requirements for in-house IVD CDx under the National Pathology Accreditation Scheme.
The CDx framework does not restrict the clinical practice of health practitioners prescribing or recommending treatments or diagnostic tests, so this is also not covered in this guidance.
This guidance document does not address the requirements for clinical trials involving IVDs, medicines, or biologicals.
Refer to the clinical trial handbook for guidance on clinical trials.
Reimbursement processes are also not within the scope of this guidance document.
CDx continue to evolve. We will monitor developments, review the implementation of the framework in 12 months’ time and update this guidance as necessary.
IVD companion diagnostics (CDx)
An IVD companion diagnostic (CDx) is an IVD medical device which provides information that is essential for the safe and effective use of a corresponding medicine or biological.
The term ‘IVD companion diagnostic’ has been defined in the Therapeutic Goods (Medical Devices) Regulations 2002 (the Medical Devices Regulations) as:
- It is an IVD medical device or an in-house IVD medical device; and
- It is intended by its manufacturer to be used for the examination of a specimen from the body of an individual:
- to identify whether the individual would be likely to benefit from the use of a particular medicine or biological; or
- to identify whether an individual is likely to be at particular risk of a serious adverse reaction to the use of a particular medicine or biological; or
- to monitor the individual’s response to the use of a particular medicine or biological; and
- It is mentioned in the product information for the medicine or the instructions for use of a biological as being essential for the safe and effective use of the corresponding medicine or biological; and
- It is not intended by the manufacturer to be used for the examination of the specimen merely to determine whether the medicine or biological is compatible with the individual (where the medicine or biological comprises blood, a blood component, cells, tissue or an organ from a donor other than the individual).
The Australian CDx regulatory framework aligns with similar international frameworks. For further details, see the United States Food and Drug Administration (FDA) guideline on Companion Diagnostics; and the European requirements under Clause 7 Article 2 of the EU In Vitro Diagnostics Regulation (IVDR), 2017-746 on Companion Diagnostics.
Medicines or biological indications that require CDx testing
Consistent with the regulatory definition, a CDx is an IVD that is essential for the safe and effective use of a corresponding medicine or biological, where both:
- the Product Information (PI) for the medicine or Instructions for Use (IFU) for the biological states that CDx testing is essential for the safe and effective use of the medicine or biological, and
- the CDx claims that it is intended for such testing, to enable the use of the relevant medicine or biological.
The PI for a medicine or IFU for a biological does not need to use the specific wording above for its meaning to be that CDx testing is essential for its safe and effective use, and the CDx testing may only relate to some, and not all uses (indications) of the medicine or biological.
Note
The term “a particular medicine or biological” can be interpreted as a “specific group of medicine or biological products”.
For example, if the intent of an IVD medical device or in-house IVD medical device is to provide information relating to the use of a specific class of medicines with a similar mechanism of action, rather than an individually named medicine or biological product, then the definition of an IVD companion diagnostic would still apply.
TGA CDx process
The CDx process aims to ensure the analytical and clinical performance of CDx are appropriate for use in conjunction with the relevant medicine or biological.
Figure 1 below presents a concept diagram of the TGA CDx evaluation process for medicine, biological, and device sponsors. A CDx testing identification guide for new medicine or biological indications is presented in Figure 2, and one for devices claiming CDx status is presented in Figure 3.
The CDx testing identification guides aim to help medicine or biological sponsors identify whether a proposed indication requires CDx testing, and if so, the companion testing plan requirements for their application.
The guides also help device sponsors identify IVDs that are eligible for inclusion in the Australian Register of Therapeutic Goods (ARTG) under the CDx framework.
The guides also help pathology laboratories identify in-house tests that can be accredited as in-house IVD CDx and notified to the TGA.
For a medicine or biological sponsor, the TGA CDx evaluation process starts in the pre-submission phase. When a medicine or biological sponsor intends to register a new indication, they are encouraged to use the medicine CDx identification guide in Figure 2 to decide whether the requested indication needs CDx testing. If so, they will need to decide on a companion testing plan option listed in Figure 4 and include it in Module 6 of the dossier. Module 6 will be new in the Australian eCTD specification version 3.2. In the interim, sponsors may use their preferred location; usually module 1.0.3 or 3.2.R until eCTD 3.2 is live.
Communicate the selected dossier location for the CDx information in your cover letter.
If the sponsor is unsure whether the new indication needs a companion testing plan, a pre-submission meeting with the Prescription Medicines Authorisation Branch is recommended.
At the preliminary assessment phase, the TGA will assess whether the requested indication requires CDx testing.
If it is concluded that no CDx testing is required, then the medicine application proceeds without a companion testing component evaluation.
If it is concluded that the requested indication requires CDx testing, information to allow TGA to evaluate the IVD used in generating the pivotal data (the clinical trial assay; see Clinical trial assay evaluation) and a companion testing plan (see Companion testing plan) will need to be in the dossier. The TGA will contact the sponsor prior to milestone 2 (for medicine/biological applications) if our assessment differs from the sponsor’s assessment, or if the TGA requires further information regarding a companion testing plan or the clinical trial testing.
During the medicine evaluation, if CDx testing is required, the device aspects will be assessed as a component of the medicine evaluation, similar to a risk management plan. This will involve the assessment of the scientific validity, and analytical and clinical performance of the clinical trial assay. In addition, if option 4 of the companion testing plan in Figure 4 is selected, the IVD proposed by the sponsor under option 4 to be used with the medicine or biological will be also reviewed by the TGA within the medicine or biological application for:
- Scientific validity, and analytical and clinical performance, and
- Comparability to the clinical trial assay to establish clinical utility.
The medicine or biological decision delegate incorporates the advice from the CDx component evaluation into their decision about the registration of the proposed new indication. If approved, information is published to assist stakeholders in identifying that this indication requires CDx testing. Section 4.2 of the Australian Product Information (PI) should contain a ‘flag’ phrase similar to the following, where square brackets indicate placeholder text or explanatory notes:
“Purpose of testing e.g. patient selection
Prior to the use of [medicine] to treat [indication], [test outcome] [must be established/is essential]. Testing used in clinical practice should be adequately comparable to the testing used in clinical studies (refer to section 5.1 Pharmacodynamic properties – clinical trials) [or refer to the section of the Product Information that describes the clinical studies]. A list of IVD companion diagnostic (CDx) tests is available on the TGA website.”
The absence of a ‘flag’ phrase from a PI document does not necessarily mean the medicine or biological does not require CDx testing for any of its indications. The ‘flag’ phrase is intended to provide stakeholders with an easy way to identify new indications that involve CDx testing. The TGA does not require addition of the ‘flag’ phrase to PIs for existing indications.
If the indication is approved, and the medicine or biological sponsor is using option 4 of the companion testing plan (Figure 4), a condition of registration will be put in place post-approval to allow the medicine or biological sponsor to commit to adhering to their plan and keeping the TGA informed in a timely manner of clinically significant changes. Sponsors are expected to support changes with data where appropriate. Applications with data should be provided as a type H submission to enable evaluation of the data. The condition of registration can be removed if the sponsor’s companion testing plan is changed to option 1, 2 or 3.
For device sponsors, the first step is to consider, if applying to include a new IVD in the ARTG whether it is intended for use as a CDx for corresponding medicine or biological products. The TGA assesses CDx device applications for scientific validity, analytical performance and clinical performance, as for any IVD application. Proof of clinical utility comes from the pivotal trials, so the IVD must establish comparability to the testing used in those trials. Information from the medicine or biological companion testing plan or the CDx component evaluation of the clinical trial assay can be used as additional evidence to inform the assessment of the IVD application.
Pathology laboratories who wish to seek accreditation of an IVD that is intended for use as a CDx for corresponding medicine or biological products must arrange to have their in-house IVD CDx to be accredited under the National Pathology Accreditation Scheme and to notify the TGA under a separate process. In-house IVDs do not become CDx, unless they make claims of being essential for safe and effective use of the corresponding medicine or biological. For more information on in-house IVD CDx, refer to the in-house section of this guidance.
The TGA publishes approved CDx on our TGA CDx List. This is a list of the approved CDx IVDs that are available in Australia for each newly registered medicine or biological indication that requires such testing. The TGA will include accredited in-house IVD CDx in the List soon.
There may be up to a month lag between an approval or notification and an update of the list entry.
Figure 1. Concept diagram of the TGA CDx evaluation process
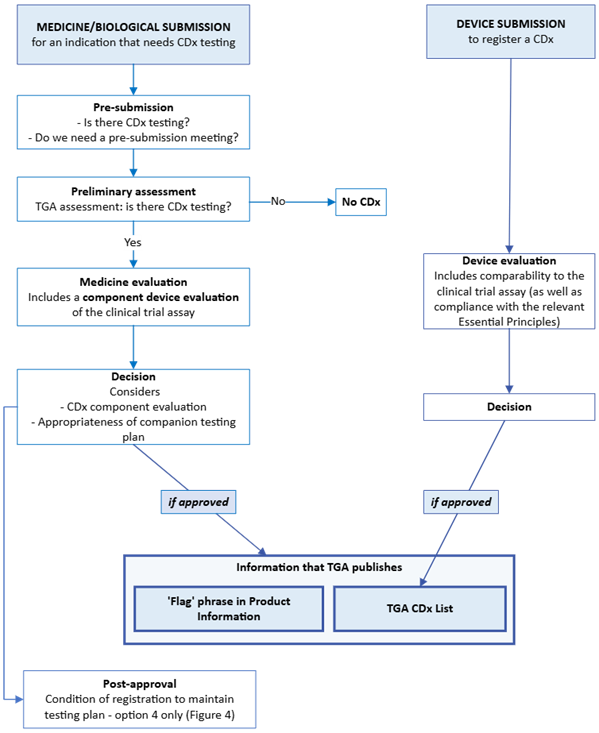
{kind=link}
Figure 1. shows a flowchart illustrating two parallel regulatory pathways:
- Left side - MEDICINE/BIOLOGICAL SUBMISSION for an indication that needs CDx testing:
- Begins with Pre-submission assessment of CDx testing requirements
- Moves to Preliminary assessment by TGA to determine if CDx testingis required
- If "Yes," proceeds to Medicine evaluation (includes component device evaluation of clinical trial assay)
- Leads to Decision stage considering CDx component evaluation and appropriateness of companion testing plan
- If approved, information is published by TGA
- Ends with Post-approval conditions to maintain testing plan (option 4 only, Figure 4)
- Right side - DEVICE SUBMISSION to register a CDx:
- Leads directly to Device evaluation (includes comparability to clinical trial assay)
- Proceeds to Decision stage
- If approved, information is published by TGA
Both pathways converge at a box showing "Information that TGA publishes," which includes a "Flag phrase in Product Information" and "TGA CDx List."
The diagram uses directional arrows to show process flow, with decision points clearly marked by "Yes," "No," and "If approved" indicators.
Figure 2. CDx identification guide for new medicine or biological indications
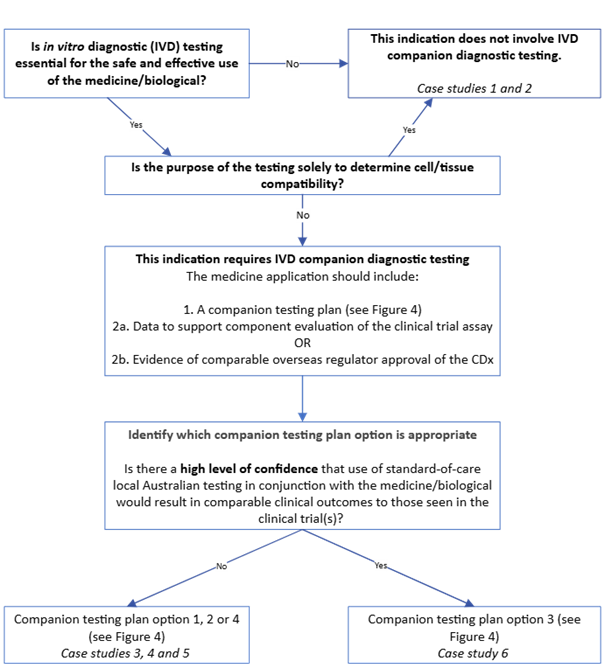
{kind=link}
Figure 2. shows a decision flowchart for determining companion diagnostic testing requirements for medicines/biologicals. The flowchart includes:
- Starting question in box: "Is in vitro diagnostic (IVD) testing essential for the safe and effective use of the medicine/biological?"
- "No" pathway arrow leads to: "This indication does not involve IVD companion diagnostic testing. Case studies 1 and 2"
- "Yes" pathway arrow leads to next decision point.
- Second question: "Is the purpose of the testing solely to determine cell/tissue compatibility?"
- "Yes" pathway arrow connects back to "This indication does not involve IVD companion diagnostic testing"
- "No" pathway arrow continues downward.
- Text box: "This indication requires IVD companion diagnostic testing" followed by application requirements:
- "1. A companion testing plan (see Figure 4)"
- "2a. Data to support component evaluation of the clinical trial assay OR"
- "2b. Evidence of comparable overseas regulator approval of the CDx“.
- Next step: "Identify which companion testing plan option is appropriate".
- Assessment question: "Is there a high level of confidence that use of standard-of-care local Australian testing in conjunction with the medicine/biological would result in comparable clinical outcomes to those seen in the clinical trial(s)?".
- "No" pathway arrow leads to: "Companion testing plan option 1, 2 or 4 (see Figure 4) Case studies 3, 4 and 5"
- "Yes" pathway arrow leads to: "Companion testing plan option 3 (see Figure 4) Case study 6".
The flowchart uses directional arrows to indicate "Yes" and "No" paths, and boxes for decision points and outcomes.
Figure 3. CDx identification guide for devices claiming CDx status
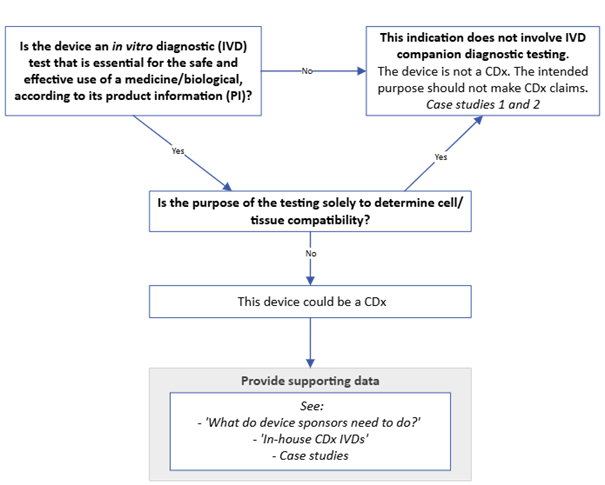
{kind=link}
Figure 3. shows a decision flowchart for determining if a device qualifies as a companion diagnostic (CDx). The flowchart includes:
- Starting question in box: "Is the device an in vitro diagnostic (IVD) test that is essential for the safe and effective use of a medicine/biological, according to its product information (PI)?"
- "No" pathway arrow leads to: "This indication does not involve IVD companion diagnostic testing. The device is not a CDx. The intended purpose should not make CDx claims. Case studies 1 and 2"
- "Yes" pathway arrow leads to next decision point.
- Second question in box: "Is the purpose of the testing solely to determine cell/tissue compatibility?"
- "Yes" pathway arrow connects back to "This indication does not involve IVD companion diagnostic testing..."
- "No" pathway arrow continues downward.
- Box stating: "This device could be a CDx".
- Box below with supporting information: "Provide supporting data" followed by:
- "See:"
- "'What do device sponsors need to do?'"
- "'In-house CDx IVDs'"
- "Case studies".
The flowchart uses directional arrows to indicate "Yes" and "No" paths, and boxes for decision points and outcomes.
Figure 4. Companion testing plan options
The companion testing plan should describe how patients in Australia would be able to access adequate CDx testing to allow the medicine or biological to be used safely and effectively for the proposed indication, if it is approved.
Give your plan a date and a ‘version number’ or other identifier of your choosing, to enable version control.
Only one adequate test needs to be identified.
Choose one of the following companion testing plan options:
Option 1 An application for TGA registration of a corresponding CDx is planned or underway. |
|---|
Provide:
|
Option 2 An Australian pathology laboratory has applied, or plans to apply, for their corresponding in-house CDx to be accredited under the National Pathology Accreditation Scheme. |
|---|
Provide:
|
Option 3 Standard testing in Australia is anticipated to predict similar clinical outcomes to the pivotal clinical trials supporting the proposed medicine or biological indication. |
|---|
Provide:
A “standard” test should be available to any patient in Australia requiring such testing. Beyond this consideration, the TGA does not have an official definition or set list of assays that are considered “standard”. We encourage sponsors to contact StreamlinedSubmissions@health.gov.au or IVDs@health.gov.au if they have questions about a particular test. |
Option 4 None of the above. |
|---|
Provide:
|
How are CDx regulated?
Classification
Classification rule 1.3(fa) in Schedule 2A of the Medical Devices Regulations, specifies that all CDx are class 3 IVDs or class 3 in-house IVDs.
Separate applications for each CDx
The CDx regulations were introduced on 1 February 2020. Under the CDx regulatory framework, each CDx will require a separate application and need to be included in the ARTG separately with a Unique Product Identifier (UPI) that is unique and determined by the CDx manufacturer. This differs from the requirements for many other class 3 IVD medical devices for which a single ARTG inclusion can cover a range of devices with the same Global Medical Device Nomenclature (GMDN) code. Device sponsors are required to indicate in the application form that the application is for a CDx. This allows the TGA to align the device assessments with the associated medicine or biological evaluation. It also ensures that devices are appropriately assessed in the context of their intended purpose and level of risk. The UPI (and a functional description) for the device will need to be entered in the application form and is generally a combination of words, numbers, symbols, or letters assigned by the manufacturer to uniquely identify an individual IVD. The UPI is directly reflected on the ARTG certificate and in the public summary for the ARTG entry, therefore, it is strongly encouraged that the name of the device and catalogue number are included in the UPI.
Application audit
The TGA must select an application to include a CDx in the ARTG for a mandatory application audit with an associated assessment fee, unless the application is supported by a conformity assessment document issued by the TGA or by one of the following comparable overseas regulators (Regulation 5.3 of the Medical Devices Regulations):
- the European IVD Regulations (2017/746)
- Premarket Approval issued by United States Food and Drug Administration
- Japanese Pharmaceutical and Medical Devices Agency
- Singapore Health Sciences Authority
- Medical Device License issued by Health Canada.
However, we may still select any application supported by comparable overseas regulator documents for a non-mandatory application audit, which will not incur an assessment fee.
For an application selected for audit, the TGA will request the sponsor to submit the information required to conduct the application audit. For application audits, sponsors are typically expected to submit all data to demonstrate conformity to the Essential Principles of safety and performance through the preparation and submission of a technical file that shows how the IVD was designed, developed, and manufactured. Further details of the technical file required for an application audit can be found in Application audit (technical file review) of IVD medical device applications.
All inclusions of CDx in the ARTG are subject to automatic conditions which are imposed at the time a device is included in the ARTG. There may be additional non-standard conditions of inclusion imposed, if necessary.
Concurrent submission for CDx and the corresponding medicine or biological
IVDs are often developed by different organisations to those involved in the development of medicines or biologicals.
The TGA encourages sponsors to submit a concurrent device application for a CDx for each corresponding medicine or biological indication requiring CDx testing.
However, this may not necessarily be possible within the same timeframe. CDx testing may also rely on the use of in-house IVD CDx. All these factors may make it difficult to submit concurrent applications for a CDx and for the relevant indication for use of a corresponding medicine or biological. While strongly encouraged, concurrent submission and assessment of these applications is not mandatory.
However, a CDx application is predicated on the existence of the corresponding medicine or biological indication for use. A submission for ARTG inclusion of an IVD with an IFU that makes CDx claims should not be made unless the relevant indication for use of a corresponding medicine or biological is approved or is under concurrent review by the TGA.
TGA CDx List
The TGA publishes a CDx List of assays approved for supply in Australia under the CDx framework, along with the corresponding medicine or biological indication for which CDx testing is required. The list identifies commercially supplied CDx that the TGA has assessed and included in the ARTG. In-house IVD CDx that have been accredited under the National Pathology Accreditation Scheme and notified to the TGA will be added to the CDx List soon.
The CDx List does not have formal regulatory status, but serves as a communication tool to assist clinicians, laboratories and other stakeholders to identify CDx tests that have been approved for supply in Australia and been assessed for adequate comparability to testing used in the relevant pivotal medicine or biological studies.
The published list will include all commercial CDx approved after introduction of the CDx framework and new medicine or biological indications that require CDx testing where no CDx is available in Australia. However, the list may not include information on all medicine or biological indications that require CDx testing, but that were approved prior to introduction of the CDx framework.
What medicine or biological sponsors need to do
At the time of submission for a new indication for a medicine or biological (regardless of whether the active substance is new or already registered), the sponsor of the medicine or biological should use the CDx identification guide for new medicine/biological indications in Figure 2 to determine whether the indication requires CDx testing, and if so, the most appropriate companion testing plan. If the sponsor is not sure, we recommend consulting the TGA by requesting a pre-submission meeting with the Prescription Medicines Authorisation Branch.
When considering whether testing is ‘essential’ to a medicine or biological indication, sponsors should consider the consequences of false positives, false negatives, test failures and no results of the CDx required for their indication. Tests that are intended by the manufacturer to be used for the examination of the specimen merely to determine whether the medicine or biological is compatible with the individual (where the medicine or biological comprises blood, a blood component, cells, tissue or an organ from a donor other than the individual) are specifically excluded by the legislation. Therefore, if the sole purpose of the test is to determine cell/tissue compatibility, the device is not considered a CDx. This applies, for example, to IVDs that solely determine ABO compatibility for blood transfusions or HLA matching for organ transplantation.
For medicine or biological indications that require CDx testing, the medicine or biological submission must include:
- Data to support evaluation of the scientific validity, analytical performance and clinical performance of the clinical trial assay (see Clinical trial assay evaluation), and
- A companion testing plan identifying an IVD that is considered suitable by the sponsor for companion testing in Australian clinical practice (see Companion testing plan).
The testing identified in the companion testing plan could be:
- the original clinical trial assay, or
- a different IVD than the clinical trial assay (i.e., a ‘subsequent’ CDx).
As it is often not possible for actual testing in clinical practice to be identical to the testing carried out in a clinical trial, the Australian CDx framework does not require a one-to-one relationship between an indication and a single proprietary CDx test. Instead, it focusses on the core characteristics of the clinical trial assay that a subsequent CDx would need to emulate comparably for it to adequately guide safe and effective use of the medicine or biological. The ‘companionship’ exists between an indication for use of a particular medicine or biological and those core characteristics of testing, rather than an individual test.
To identify which companion testing plan option is appropriate, the sponsor should consider whether there is a high level of confidence that the use of standard local Australian testing in conjunction with the medicine or biological would result in comparable clinical outcomes to those seen in the clinical trials.
Examples of factors that could increase confidence:
- The test is well-established in Australia with widespread and extensive experience of clinical usage and pathology guidance, such that inter-reader variability is very unlikely to be clinically meaningful.
- Standardised reference materials (calibrators and controls) are readily available to ensure consistent performance.
- Inter-laboratory variability is acceptable and very unlikely to be clinically meaningful.
Examples of factors that could reduce confidence:
- The test is novel (methodology or target/outcome).
- The test itself is not novel but was used in a novel way in clinical trial testing.
- The test involves a methodology known, or anticipated to have, high variability in performance.
- The test is a member of a class of tests within which there is clinically relevant heterogeneity.
Clinical evaluation
During review of a new indication that requires CDx testing, the TGA will evaluate the clinical evidence supporting the use of the clinical trial assay in the pivotal study. As outlined on our Meeting clinical evidence requirements for in-vitro diagnostic (IVD) medical devices page, this involves consideration of the analytical performance and clinical performance of the test, as well as its scientific validity and clinical utility (i.e. the pivotal clinical studies). The evaluation of the clinical trial assay will consider its core characteristics, to support assessment of any subsequent CDx applications that may follow.
The data required to support the clinical trial assay evaluation depends on the nature of the testing and the assay used, and the companion testing plan (see below).
For well-established testing using a commercial assay that is approved by a comparable overseas regulator, reference to the name of the test and a cross-reference to the overseas approval or published validation data can be sufficient.
Where a bespoke clinical trial assay with no existing regulatory approvals has been used in the pivotal clinical studies, provide more detailed data to support TGA evaluation, including:
- Device regulatory history, including any international regulatory approvals, rejections or withdrawals
- Design and manufacturing information
- Instructions for use (if available)
- Clinical performance data
- Analytical performance data, including validation of any controls/calibrators/reference materials or internal standards utilised
- Risk analysis
- Stability data.
If you are unsure what data should be included, contact StreamlinedSubmissions@health.gov.au and IVDs@health.gov.au for advice.
Companion testing plan
A companion testing plan (see Figure 4) is information provided by the sponsor of a medicine or biological product, relating to an indication that requires testing with a CDx, nominating how such testing will be available to Australian patients. The purpose of the plan is to provide reassurance that there will be access to at least one adequate CDx and to therefore ensure that Australian patients can be treated for that indication safely and effectively. All applications for registration of a new medicine or biological indication that requires CDx testing must include a companion testing plan. The plan should include a date and version control number (of the sponsor’s choice of format), for version control purposes.
The plan only needs to identify one CDx that the TGA considers adequate for that particular medicine or biological indication. The companion testing plan is not a comprehensive description of all possible CDx that are available in Australia at the time of medicine indication registration (or over subsequent time).
Making a companion testing plan consists of selecting one of four general options (Figure 4).
If the medicine sponsor is aware of a concurrent application for inclusion of a corresponding CDx in the ARTG (or notification of an in-house IVD CDx), the companion testing plan can consist of a cross-reference to the relevant details (Figure 4; option 1). Where a medicine's sponsor expects the testing to be conducted by an in-house IVD CDx, the companion testing plan could state this and provide relevant information (Figure 4; option 2). Where the Sponsor considers that standard Australian testing is adequate for CDx use with the medicine, they should provide detailed rationale for this (Figure 4; option 3). If none of the above apply, and the Sponsor intends on alternative arrangements, for example, ‘send-away’ off-shore testing, full details of the intended testing need to be made available for TGA review (Figure 4; option 4).
In including ‘option 4,’ the companion testing plan provides a mechanism for the TGA to evaluate the performance and validity of CDx testing, even when there is no concurrent application for inclusion of a CDx in the ARTG (or notification of an in-house IVD CDx). This approach is to recognise and acknowledge that there may be barriers to bringing a CDx to the Australian market for local supply, and Australian samples may have to be sent for testing internationally. While this is not preferred, the companion testing plan provides a mechanism for the TGA to appraise such testing until the registration or notification of a local testing option is possible.
Note
The TGA considers sending of samples to an appropriately accredited overseas testing facility acceptable only if development of onshore testing with an ARTG-included or in-house (notified) CDx test is unavailable.
The TGA encourages device and medicine or biological sponsors to work together to identify these gaps in the Australian CDx market and expedite development of Australian based CDx.
If ‘option 4’ is selected, the medicine/biological sponsor will need to provide:
- Information about test availability and accreditation of destination laboratories
- Device history, including any international regulatory approvals, rejections or withdrawals
- Design and manufacturing information
- Clinical performance data
- Analytical performance data, including validation of any controls/calibrators/reference materials or internal standards utilised
- Risk analysis
- Stability data
- Evidence of comparability to the clinical trial testing
The requirements for evidence of comparability are the same as would be required for Australian registration of a subsequent CDx.
Note
A medicine or biological indication that requires CDx testing can be approved without a corresponding CDx test being on the ARTG (or notified to the TGA as an in-house IVD), if an adequate companion testing plan is in place.
However, a commercial CDx must be included in the ARTG (or an in-house CDx must be accredited under the National Pathology Accreditation Scheme) before the CDx can be legally supplied in Australia.
Post-approval actions
When a medicine or biological indication that requires CDx testing is approved, it will be published on the TGA CDx List along with the testing that is available in Australia for use with it.
For medicine or biological indications with companion testing plans that use option 4, the entry in the List will reflect the lack of on-shore testing options, and it can be understood that there is scope for development of an Australian test. Additionally, a condition of registration will be that the medicine or biological sponsor will adhere to their companion testing plan (including the plan date and version; for version control) and will notify the TGA if there are any substantial changes to the companion testing plan. While “substantial” has not been specifically defined for this purpose, examples of changes that the TGA would need to be notified of are:
- The test is subject to supply interruption
- The test is subject to regulatory action such as cancellation in another jurisdiction
- A change to the testing methodology, core characteristics or location such that the sponsor considers that clinically meaningful impacts on the results might occur.
For medicine or biological indications with companion testing plans that use option 3, conditions of registration relating to a companion testing plan are not needed.
For medicine or biological indications with companion testing plans that use option 1 or 2, a condition of registration relating to the companion testing plan is unlikely to be needed if a related CDx device application (or accreditation) is close to completion at the time of the medicine or biological registration decision.
In an approved indication is initially subject to a companion testing plan-related condition of registration, and a corresponding CDx is subsequently approved by the TGA (or notified to the TGA, under the in-house IVD framework), the condition of registration can be removed without requiring additional evaluation of data. This can be done by a TGA delegate of their own volition or at the request of the sponsor.
Data to support changes to a companion testing plan that uses option 4 should be submitted by the medicine or biological sponsor through a type H application, as these data require evaluation.
It is conceivable that available testing in Australia may become unavailable: for example if serious concerns about standard testing arose, or if all existing (approved and notified) CDx for an existing approved medicine or biological indication became unavailable in Australia. This is not anticipated to be a common instance but could be managed by revisiting the companion testing plan for an indication if such a situation arose.
Product Information (PI) or Instructions For Use (IFU) for the medicine or biological
The PI for a medicine or the IFU (for a biological) should include a CDx ‘flag’ statement for all new indications that require CDx testing, similar to the following, amended as clinically appropriate:
“Patient selection
Prior to the use of [medicine] to treat [indication], [test outcome] must be established [/is essential]. Use a validated test that is adequately comparable to the testing used in clinical studies (see [section of the document that describes the clinical study e.g. 5.1]). A list of IVD companion diagnostic (CDx) tests that have been assessed for comparability is available on the TGA website."
The section of the document that describes the clinical trial (e.g. section 5.1 Pharmacodynamic properties – Clinical trials) should include a short statement identifying the testing used in the study. For example, “Patients were eligible to enrol based on central testing using the [BRAND] assay” or “Patients with an EGFR or ALK mutation according to local testing were excluded from the study.”
In instances where all pivotal clinical studies relating to any approved indication used standard local testing for enrolment, the sentence about adequate comparability can be omitted as it is of limited clinical meaningfulness.
For medicines, it is recommended that a CDx flag statement be included as a subheading in section 4.2 of the PI (Dose and method of administration). A single statement listing all relevant indications is preferred rather than a separate statement/new paragraph per indication.
The TGA does not intend to retrospectively require existing approved medicine or biological indications that require CDx testing to add a CDx flag phrase to their PI. Existing PIs for such medicines may contain various testing-related terminology (such as “validated test”). The flexibility in the legislation wording allows those older indications to still be considered ‘indications requiring companion testing’ without needing a specific CDx ‘flag’ phrase to be in the PI. However, as the intention of the new flag phrase is to assist stakeholders to identify the medicine indications that correlate with registered CDx devices or notified accredited tests, we encourage sponsors of older indications to alter their PIs to add a similar phrase. This can be done at a convenient time within an existing submission and is encouraged but not mandated.
The TGA does not plan to require specific references to CDx testing in the wording of the indication to allow for clinically appropriate flexibility in real-world usage. Selection of patients that match the clinical indication description (including selection of clinical tools such as IVD diagnostic tests with which to do so) is within the remit of clinical practice, which TGA does not regulate. The intention of the Australian CDx framework is to provide additional information that stakeholders can use in their decision-making, including identifying where the TGA has made a specific assessment of comparability between tests.
What device sponsors need to do
Sponsors of an IVD test seeking inclusion as a CDx in the ARTG must submit an IVD medical device application while the medicine or biological application is being considered, or at any time after approval of the medicine or biological. Instructions for the submission of IVD medical device applications can be found on the TGA website at: Medical device inclusion process.
The IVD medical device application must include data to demonstrate minimum applicable conformity assessment procedures and compliance with the relevant Essential Principles:
- Device history, including any international approvals, rejections or withdrawals.
- Design and manufacturing information
- Instructions for use (IFU)
- Clinical performance
- Clinical utility, as applicable
- Analytical performance, including validation of any controls/calibrators/reference materials or internal standards utilised.
- Risk analysis
- Stability data.
The intended purpose in the application form for a CDx must exactly match the intended purpose in the IFU, as this is the source of the intended purpose for the ARTG certificate.
When considering whether testing is ‘essential’ to a medicine or biological indication, sponsors should consider the consequences of false positives, false negatives, test failures and no results of the CDx required for their indication. Tests that are intended by the manufacturer to be used for the examination of the specimen merely to determine whether the medicine or biological is compatible with the individual (where the medicine or biological comprises blood, a blood component, cells, tissue or an organ from a donor other than the individual) are specifically excluded by the legislation. Therefore, if the sole purpose of the test is to determine cell/tissue compatibility, the device is not considered a CDx. This applies, for example, to IVDs that solely determine ABO compatibility for blood transfusions or HLA matching for organ transplantation.
A sponsor seeking TGA IVD registration of a CDx (or laboratories seeking accreditation of a CDx) in relation to a medicine or biological indication will normally need to provide direct clinical comparability data between their test and the clinical trial assay.
For further details on these requirements, refer to the following sections;
- Clinical performance requirements,
- Clinical utility for subsequent CDx,
- Analytical performance requirements,
- Clinical evidence for in-house CDx IVDs.
If the sponsor can establish, based on robust non-clinical data, that there is a very high level of confidence that use of their test as a CDx in conjunction with the medicine or biological would result in comparable clinical outcomes to those seen in the clinical trials, indirect clinical comparability data may be acceptable.
We define direct and indirect clinical comparability data the same way whether discussing commercial or in-house IVDs. See Clinical evidence for in-house IVD CDx.
Instructions for use (IFU)
The TGA uses the term ‘CDx claims’ to refer to statements about the IVD’s intended purpose:
- in the selection of patients for treatment with a particular medicine or biological; or
- in the monitoring of patients who are being treated with a particular medicine or biological; or
- in both selection and monitoring of treatment with a particular medicine or biological.
CDx claims in the IFU are expected to reference the International Non-proprietary Name (INN) of the corresponding medicine or biological.
IVDs that match the applicable core characteristics (of the pivotal clinical trial testing that supported registration of the relevant indication for use for the corresponding medicine or biological) will be allowed to make CDx claims in the IFU and be included in the ARTG as CDx for the relevant use of the corresponding medicine or biological. Approved CDx IVDs will be published on the TGA CDx List.
Ambiguous claims in the IFU regarding whether a product is intended for use as a CDx are not acceptable.
Examples of intended use statements consistent with CDx claims:
- The primary use of the (IVD name) is the detection of the BRAF V600 mutations in DNA extracted from formalin-fixed, paraffin-embedded human melanoma and papillary thyroid carcinoma (PTC) tissue. In melanoma, it is intended to be used as an aid in selecting patients whose tumours carry BRAF V600 mutations, for treatment either with the medicine name alone, or for treatment with medicine 1 in combination with medicine 2.
- PD-L1 expression in tumour cell (TC) membrane as detected by (IVD name) in NSCLC is indicated as an aid in identifying patients for treatment with the medicine.
- The assay (In-house IVD) is intended to be used as a companion diagnostic to be ordered by Australian medical oncologists to identify ovarian cancer patients with homologous recombination deficiency (HRD), who may benefit from treatment with the medicine name in combination with standard therapy as a maintenance therapy following first line chemotherapy.
Note
An IVD sponsor cannot register a CDx unless the corresponding medicine or biological has been approved for the relevant use in Australia, or there is a concurrent application for approval of the relevant indication for use of the corresponding medicine or biological.
The TGA does not require the decision dates for the CDx and medicine or biological applications to align.
Transitional arrangements
Transitional arrangements are in place for device sponsors who had CDx included in the ARTG as class 2 or 3 IVDs before 1 February 2020 and for pathology laboratories with in-house IVDs. Sponsors can continue to supply those CDx until 31 December 2028. However, a new application that complies with the amended Regulations must be made before the transition end date to continue supply after 31 December 2028.
These include CDx IVDs that:
- are included in the ARTG; or
- are not included in the ARTG but are covered by a current conformity assessment certificate issued by the TGA; or
- are in-house IVDs that are Class 1, Class 2, or Class 3 in-house IVDs (noting that CDx will be Class 3 under the new Regulations).
CDx IVDs that meet these criteria are allowed to continue supply as class 3 IVDs until the transition end date of 31 December 2028.
Clinical utility for subsequent CDx
A subsequent CDx is an IVD that is different to the clinical trial assay but is intended for the same medicine and indication. Evidence of clinical utility must be provided for a subsequent CDx. This involves a combination of clinical and analytical performance studies. The following sections regarding CDx clinical and analytical performance requirements should be read in conjunction with the guidance: clinical evidence guidelines supplement: In vitro diagnostic (IVD) medical devices.
Applicants may use bridging and comparability studies to demonstrate comparable analytical and clinical performance between the original CDx and the subsequent CDx. This would enable the pivotal studies that supported the indication approval of the original CDx to demonstrate the clinical utility for approval of the subsequent CDx.
Alternatively, a comparative clinical study with adequate statistical methodology and appropriate clinical endpoints could be performed establishing a direct link between the results of the subsequent CDx and patient outcomes for that medicine indication. The data would need to demonstrate that the clinical outcomes were not significantly different with use of the subsequent CDx to those that were obtained using the clinical trial assay in the original pivotal studies. Adequate clinical justification would be required for the statistical parameters in the study design, which should be pre-specified, including power and acceptance criteria (margins for concluding equivalence or non-inferiority).
Clinical performance requirements
There are two ways that a CDx can be made available for supply in Australia:
- Application for inclusion of the CDx in the ARTG
- Accreditation of an in-house CDx under the National Pathology Accreditation Scheme. The pathology laboratory also needs to notify the TGA that it is supplying an in-house CDx.
If supply of a test in Australia through one of these mechanisms is not feasible, overseas testing may be allowed to be part of the companion testing plan for the medicine or biological.
Table 1 describes the clinical performance requirements for an IVD that is intended to be used as a CDx, based on the way the IVD was developed with relation to the original CDx (the pivotal clinical trial assay).
| Scenario | 1 | 2 | 3 |
|---|---|---|---|
| Proposed CDx for inclusion in the ARTG is the original CDx (the same as the clinical trial assay) | Proposed CDx for inclusion in the ARTG is a subsequent CDx being developed commercially | A laboratory seeks to develop their own in-house IVD for use as a proposed CDx | |
| Requirements | Clinical studies need to be well-designed. Aspects such as the prevalence of the target analyte, the statistical confidence and the adequate characterisation of all samples included in the study must be considered. | ||
Substantial equivalence (clinical comparability) of the subsequent CDx with the original CDx must be demonstrated. This will be contextualised by the core characteristics of the pivotal clinical trial testing, and could be based on:
The TGA may consider other parameters when assessing whether there is substantial equivalence between a subsequent CDx and a clinical trial assay, such as the composition and nature of the tests. Substantial equivalence could be demonstrated by indirect evidence of clinical comparability if it is robust enough to establish a very high level of confidence that use of this test as a CDx in conjunction with the medicine or biological would result in comparable clinical outcomes to those seen in the clinical trials. | |||
Refer to requirements for in-house IVD CDx. In-house IVD CDx must also comply with the validation requirements set out in the National Pathology Accreditation Advisory Council (NPAAC) standard. | |||
Analytical performance requirements
Analytical performance studies will be expected to include full validation of the following:
- Specimen and assay stability (storage and transport)
- Specimen equivalence (for devices which intend to use more than one type of specimen)
- Sensitivity (limit of detection, limit of blank, limit of quantitation, as appropriate)
- Assay cut-off, or decision points for the assay, if applicable
- Specificity (interference, cross-reactivity, inclusivity, any Hook Effect or prozoning)
- Linearity or measuring range
- Precision and accuracy
- Quality control material (including reference materials or internal standards utilised)
- If it is a semi- or quantitative assay, calibration material must be available and expressed in acceptable or convertible units of measurement.
Full details of all studies conducted including experimental design and individual results for each individual sample utilised are required and summary details will not be accepted.
For more information on the requirements of detailed information, refer to depth of information to be provided.
Changes to CDx included in the ARTG
The sponsors of all CDx included in the ARTG can apply to the TGA if they need to change any details of the device by completing the Device Change Request form. Changes could include:
- A change in name (UPI) of the CDx
- A change to the intended purpose
- Examples could include a change to the target or biomarker detected, addition of other targets or biomarkers to be detected, change in cut-off concentration or decision point for receiving the medicine or biological, expanded medicine or biological indications, addition of a new type of specimen or an addition or change to the function of the software used with the device. This list is not exhaustive but is intended to provide some examples only.
- Change to the manufacturer name or address.
Contact IVDs@health.gov.au for further information or to seek clarity on any action required for changes to the CDx included in the ARTG.
In-house IVD CDx
'In-house' IVDs are pathology tests that have been developed (or modified) within a laboratory (or laboratory network) to carry out testing on human samples, where the results are intended to assist in clinical diagnosis or be used in making decisions concerning clinical management.
Laboratories can choose to develop (or modify) an in-house test to be used as a CDx. Laboratories may work with the relevant medicine or biological product sponsor to develop an in-house IVD CDx but are not obliged to do so. In-house IVDs that are intended for use as CDx are subject to the regulatory changes (e.g. definition, classification rules) and the same transition period applies to both commercially supplied CDx and in-house IVD CDx.
The CDx framework does not restrict the clinical practice of health practitioners prescribing or recommending treatments or diagnostic tests and these clinical activities alone do not make a test into a CDx. An in-house test becomes a CDx only if a medicine or biological product needs a CDx and the laboratory intends the test to be a CDx for that medicine or biological product.
Classification
The classification rule which clarifies that all CDx are Class 3 IVDs applies equally to commercially supplied and in-house IVDs.
Notification to the TGA
Class 1-3 in-house IVDs do not require inclusion in the ARTG, however, laboratories will need to have their tests accredited and identify their in-house IVD CDx in the test list they provide to the TGA in their existing notification process.
Laboratories are required to specifically identify their CDx in the test list that they attach to their notification to the TGA. Guidance and updates on in-house IVD CDx notifications, can be found in Regulatory requirements for in-house IVDs.
Evaluation of in-house IVD CDx
The conformity assessment procedures in Schedule 3, Part 6A of the Medical Devices Regulations require laboratories who manufacture Class 1-3 in-house IVDs to be accredited by a national accreditation body, such as the National Association of Testing Authorities (NATA), as a testing laboratory and to meet the National Pathology Accreditation Advisory Council (NPAAC) standard, Requirements for development and use of in-house in vitro diagnostic medical devices (IVDs) (the NPAAC standard). Under NATA accreditation requirements, all Class 3 in-house IVDs will be evaluated for compliance with the NPAAC standard.
Under the Memorandum of Understanding (MoU) between NATA and the TGA, NATA may request TGA assistance in the technical evaluation of the analytical and clinical performance of an in-house IVD CDx, due to the need to access proprietary information regarding clinical trial assay performance characteristics and its use in establishing the medicine or biological indication. The TGA may provide technical assistance and advice on request but is not otherwise involved in NATA’s accreditation decision.
Clinical evidence for in-house IVD CDx
Laboratories must comply with the validation requirements set out in the NPAAC standard. The TGA continues to collaborate with NPAAC and provide input into the standard regarding the clinical evidence requirements for in-house IVD CDx.
Of note, as is the case for commercial CDx and detailed further below, indirect evidence supporting clinical comparability can be acceptable in some circumstances.
Typically, a laboratory that seeks accreditation of its in-house IVD for use as a CDx, is expected to provide evidence of clinical comparability (although this is not required for Option 3, Figure 4 where standard testing in Australia is anticipated to provide similar clinical outcomes).
Clinical utility of a CDx is established by the pivotal clinical trials that support regulatory approval of the corresponding medicine or biological indication. Unless the proposed CDx was used in the pivotal clinical trials, it must be established that the data hold adequate external validity for a population tested using the proposed in-house IVD CDx. This relies on establishing clinical comparability between the CDx assay used in the pivotal clinical trial and the proposed CDx. Establishing clinical comparability may be achieved through provision of direct or indirect comparability data.
Direct clinical comparability data comes from a study in which clinical samples are tested using both assays. The samples may be:
- Samples from the original pivotal clinical trial; or
- Samples from patients who are clinically similar to those who participated in the pivotal clinical trial.
Indirect clinical comparability data consist of:
- Strong evidence that analytical performance, scientific validity and clinical performance of the in-house IVD are highly comparable to the clinical trial assay; and
- Strong scientific and clinical justification that a direct clinical comparability study is not required to conclude that use of the proposed CDx will result in clinically comparable outcomes to those seen in the pivotal clinical trial.
Case studies
Below are case study examples that may help medicine, biological and IVD sponsors understand the CDx requirements. The case studies also relate to pathology laboratories with in-house IVDs.
Case study 1: A medicine indication that does not require CDx testing
NeuroPharm is the sponsor of a medicine, NeuroRelief, which is indicated for the treatment of patients with early-stage Alzheimer’s Disease. NeuroPharm provides information during the pre-submission process that the use of a test to detect amyloid beta and tau proteins in cerebrospinal fluid (CSF) in patients prior to treatment may be predictive of a better outcome.
Using the CDx identification guide for new medicine or biological indications (Figure 2), TGA and NeuroPharm determine that CSF testing is not essential for the safe and effective use of NeuroRelief. CSF testing in this clinical setting is an adjunct to the standard primary diagnostic methodology for early-stage Alzheimer’s disease, i.e. amyloid-PET scanning. Treatment with NeuroRelief can be safe and effective in the absence of CSF testing. Whilst CSF testing may be of clinical benefit, it is not essential.
NeuroPharm does not need to provide any CDx-related information (or a companion testing plan) in their medicine registration application.
Devices with an intended use for the detection of amyloid beta and tau proteins in CSF are not eligible to be included as a CDx for NeuroRelief. Such devices should instead seek inclusion or accreditation as a non-CDx IVD medical device. Inclusion or accreditation as a CDx is not appropriate.
Case study 2: An in-house IVD that is not a CDx
BioMarker Bonanza Laboratories develops an in-house IVD that is a flow cytometry assay measuring CD4 and CD8 T-cell response to cytomegalovirus (CMV) intended to aid in deciding whether a patient can cease a CMV prophylactic medication, Medicinevir. BioMarker Bonanza Laboratories is uncertain whether the in-house IVD is a CDx and seeks advice from the TGA.
Using the CDx identification guide for devices seeking CDx status (Figure 3), it is determined that the IVD test is not essential for the use of Medicinevir according to the product information (PI) for Medicinevir, because it is possible for the medicine to be used safely and effectively (i.e. as empirical treatment) in the absence of a confirmatory result for CMV. While patients may benefit from using the assay to inform decisions for clinical management, it is not essential.
BioMarker Bonanza Laboratories proceeds to seek accreditation of the assay as a Class 3 in-house IVD but not as a CDx.
Case Study 3: A medicine that requires CDx testing that is not available in Australia
BioThera is a sponsor of a medicine ‘medicinemab C’ which is indicated for the treatment of patients with haemophilia A. During treatment with medicinemab C, the serum level of ‘medicinemab C’ must be ascertained to inform dose adjustment to avoid risky exposure levels. The clinical trial used an initial-ELISA assay conducted in a centralised laboratory overseas (not in Australia). BioThera indicates that they have partnered with an IVD manufacturer (NextWave Diagnostics) to develop a commercial mabC-ELISA, however, validation for the commercial mabC-ELISA is going to be incomplete at the time of medicine approval. BioThera proposes to use the initial-ELISA assay (the clinical trial assay) as an interim measure for Australian patients until the commercial mabC-ELISA is approved and listed in the ARTG as a subsequent CDx.
During the medicine application, in the preliminary assessment stage, the TGA and BioThera determine that drug monitoring of ‘medicinemab C’ is essential for the safe and effective use of the medicine, that the test is for a novel target and that standard reference material is not readily available (i.e. the test is not well-established). There is no corresponding IVD application for a commercial CDx and it is not a mainstream pathology test. Using the CDx identification guide for new medicine or biological indications (Figure 2), BioThera identifies that the most appropriate companion testing plan option is Option 4 (see Figure 4). TGA supports the use of this option to allow access to the medicine by Australian patients, while the onshore test is being developed.
BioThera selects Option 4 in their companion testing plan (see Figure 4) and provides detailed information of the initial-ELISA assay (conducted overseas) in their medicine’s dossier, including full data related to scientific validity, analytical performance, and clinical performance of the initial-ELISA assay (its clinical utility has been demonstrated in the pivotal clinical trial). The TGA conducts an evaluation of the proposed CDx as a component of the medicine evaluation and determines that use of the initial-ELISA assay, conducted overseas, as an interim CDx testing approach is acceptable. ‘medicinemab C’ is approved with a condition of registration (refer to Figure 1) that requires BioThera to inform the TGA if CDx testing becomes unavailable or of modifications to their plan for CDx testing that result in any clinically significant changes.
No commercial mabC-ELISA has yet been included on the ARTG corresponding to this medicine indication, so an entry relating to the new medicine indication is published on the TGA’s CDx List with a CDx status of “no onshore testing available.”
Case study 4: An IVD that is a subsequent CDx
NextWave Diagnostics is a sponsor of an IVD, mabC-ELISA, which is used to monitor the concentration of ‘medicinemab C’ in patients receiving treatment for haemophilia A. mabC-ELISA was developed as a subsequent IVD (i.e. was not the assay used in the clinical trial), and it is intended to be used in clinical decision making and dose adjustment of ‘medicinemab C’, as described above in case study 3.
Using the CDx identification guide for devices seeking CDx status (Figure 3), NextWave Diagnostics confirms that testing is essential for the safe and effective use of ‘medicinemab C’ according to its PI, that the test is for a novel target and that standard reference material is not readily available (i.e. the test is not well-established).
NextWave Diagnostics submits an IVD CDx application to include the mabC-ELISA in the ARTG. NextWave Diagnostics must provide documentation in their IVD CDx application to demonstrate compliance with the Essential Principles (EPs). To comply with EPs 14 and 15(1), NextWave Diagnostics needs to provide clinical evidence including evidence to support clinical utility. NextWave Diagnostics provides a comparability or bridging study to demonstrate the clinical utility of the mabC-ELISA by establishing concordance between the mabC-ELISA and the assay used in the pivotal clinical trial for ‘medicinemab C’ (i.e. initial-ELISA). The comparability study involved re-testing (with the mabC-ELISA) of specimens used in the pivotal clinical trial, and evaluation of the clinical decision points for dose adjustment. All the evidence provided demonstrates compliance with the EPs, the mabC-ELISA IVD CDx is included in the ARTG, and published on the TGA’s CDx List.
Case study 5: A medicine that plans for CDx testing to be available in Australia using an in-house IVD
OncoPharma is the sponsor of a medicine ‘TumourX’, with a proposed indication for treating locally advanced or metastatic breast cancer with an activating XYZ1 mutation. The clinical trial assay used in the pivotal clinical trials is a commercial CDx in another jurisdiction, which the manufacturer does not wish to include in the ARTG. To support the registration of ‘TumourX’, OncoPharma arranges to support an Australian laboratory to develop a CDx under the in-house IVD pathway.
During the application to register the new medicine indication, in the preliminary assessment stage, it is determined that XYZ1 testing is essential for the safe and effective use of the medicine, that the test for the biomarker is not well-established in Australia, and that standard reference material is not readily available. There is no corresponding IVD application for a commercial CDx, however, OncoPharma intends to support an Australian laboratory to develop an in-house CDx assay. Using the CDx identification guide for new medicine or biological indications (Figure 2), OncoPharma identifies that the most appropriate companion testing plan option is Option 2 (see Figure 4).
In their application for registration of the medicine indication, OncoPharma identifies the commercial CDx that was used in the pivotal study and provides evidence supporting its validity in the form of cross-referencing an international assessment by a comparable overseas regulator.
OncoPharma selects Option 2 in their companion testing plan (see Figure 4) and provides detailed information on how they intend to support an Australian laboratory to develop an in-house CDx assay for XYZ1 that is to be accredited as a CDx for use with TumourX.
In their application for accreditation, the laboratory will need to provide clinical evidence (including evidence to support clinical utility). Clinical utility data is likely to consist of data establishing comparability with the commercial CDx IVD used in the pivotal clinical trials, therefore allowing bridging to the pivotal medicine study as evidence of clinical utility. The full assessment of the clinical evidence to support accreditation of the in-house CDx assay for XYZ1 will be conducted by NATA, the accreditation body.
In their companion testing plan, OncoPharma includes information about the laboratory with which they are in discussions (name, laboratory contact and accreditation status), and the timeline within which the laboratory expects to have completed an extension of their scope of accreditation to include XYZ1 as a CDx.
OncoPharma provide additional supporting information regarding the assay that the laboratory intends to use or adapt, including a description of the samples they intend to provide to the laboratory for clinical and analytical validation of the in-house CDx, details on how the laboratory intends to establish comparability between the new in-house CDx and the commercial CDx that was used in the original clinical study, and the actions taken to support the laboratory to complete a quality assurance program specific to XYZ1.
‘TumourX’ is approved with a condition of a registration (refer to Figure 1) that it will not be supplied until the in-house IVD CDx receives accreditation and that OncoPharma will inform the TGA if the in-house CDx becomes unavailable or if modifications to their plan for testing may result in any clinically significant impacts.
Case study 6: A medicine that uses standard local testing
PharmaGenix is a sponsor of a medicine ‘OncoCare’ which requires the detection of human epidermal growth factor receptor 2 (HER2) for first line use in patients with metastatic breast cancer. The clinical trial used an in-house IVD to identify HER2-positive (IHC 3+/ISH+) patients for treatment with ‘OncoCare’. Using the CDx identification guide for new medicine or biological indications (Figure 2), PharmaGenix has determined that HER2 testing is essential for the safe and effective use of ‘OncoCare’, it is not solely to determine cell or tissue compatibility, however, is a mainstream pathology test.
In the companion testing plan, PharmaGenix provides a justification on why the CDx testing is considered standard of care testing in Australia, i.e. a HER2-positive result (using this definition of ‘positive’) in breast cancer would generally be expected to identify the same patients regardless of the brand of test used. The justification supports that there is a high level of confidence that use of standard-of-care local Australian testing in conjunction with the relevant medicine or biological indication would result in comparable clinical outcomes to those seen in the relevant clinical trials. Therefore, the TGA agrees with PharmaGenix that local Australian testing can be used as CDx testing for this indication.
In their application for registration of the new indication for ‘OncoCare,’ PharmaGenix identifies the testing that was used in the pivotal study. Patients could be enrolled based on a HER2-positive result according to local standard testing.
PharmaGenix selects Option 3 in the companion testing plan (Figure 4) and provides it in the medicine’s dossier.
‘OncoCare’ is approved for HER-positive breast cancer. An entry is made in the TGA CDx List noting that local Australian testing is adequate for CDx testing for this medicine indication.
Case study 7: A medicine that uses a mainstream biomarker in a novel way
EndoHealth is a sponsor of a medicine ‘EndoCare>’ which requires the detection of human epidermal growth factor receptor 2 (HER2) for first line use in patients with metastatic breast cancer. The clinical trial used a commercial CDx, HER2tastic, for central confirmation of local test results, to identify ‘HER2-low’ patients for treatment with ‘EndoCare’. Using the CDx identification guide for new medicine or biological indications (Figure 2), EndoHealth has determined that HER2 testing is essential for the safe and effective use of ‘EndoCare’, it is not solely to determine cell or tissue compatibility, however, is a mainstream pathology test used in a novel way (i.e. the classification ‘HER2 low’ is not considered well-established).
In their application for registration of the new indication for ‘EndoCare,’ EndoHealth identifies the testing that was used in the pivotal study. Patients could be proposed for enrolment based on a HER2-low result according to local standard testing. However, because the classification is novel, to be eligible for enrolment, patients required central confirmation using a commercial CDx, HER2tastic. EndoHealth provides validation evidence in the form of approval documentation from a comparable overseas regulator for HER2tastic, and provides details of how the testing was conducted differently in their study with respect to the novel aspect of its use (defining HER2-low).
In their companion testing plan, EndoHealth needs to provide adequate information to describe to the TGA how patients are expected to be selected for the safe and effective use of ‘EndoCare’ in Australia, assuming ‘EndoCare’ is granted registration. Initially, EndoHealth selects Option 3 in their companion testing plan (see Figure 4). However, at a pre-submission meeting, EndoHealth and the TGA agree that in this instance, there may not be a high level of confidence that standard local testing in Australia would identify a similar population to those in the pivotal trial, because of the novel aspects to its usage.
In their subsequent submission for registration of the indication for ‘EndoCare,’ EndoHealth includes a companion testing plan in which they have selected option 2 (Figure 4).
EndoHealth includes information about a laboratory (name, laboratory contact and accreditation status) that performs standard testing for HER2, and which intends to provide HER2 testing using the HER2-low cut-off for use with the proposed medicine indication. EndoHealth provides the information that they have about the assay that the laboratory intends to use or adapt as a CDx, and information about the expected timeline for extending the scope of the laboratory’s accreditation to include HER2 testing as a CDx for ‘EndoCare’.
The laboratory applies to extend the scope of their accreditation very soon after the medicine dossier is submitted to the TGA. The laboratory includes the usual clinical evidence required, including analytical validity, clinical validity, and clinical utility. To support their CDx claim, they provide evidence of comparability between their test and HER2tastic at the novel ‘HER2-low’ cut-off, using representative clinical samples from metastatic breast cancers in the first-line setting, allowing them to bridge to the clinical utility demonstrated in the pivotal trial for the ‘Endocare’ indication.
The ‘Endocare’ indication receives a positive registration decision. A condition of registration is not required regarding CDx testing because accreditation of the in-house test is anticipated to be completed soon after the medicine approval. An entry is published on the TGA’s CDx List with the ‘Endocare’ indication, stating that a TGA-registered CDx or accredited and notified CDx is not available yet.
Once the in-house test accreditation has been completed, the list entry is updated to include the in-house CDx test.
Glossary and abbreviations
| Term | Description |
|---|---|
| Bridging study | A study required to demonstrate equivalence to the clinical trial assay, when one or more factors relevant to clinical utility of the test varies between the clinical trial assay and the subsequent CDx. For example, a variation to the subject population or a change to the assay mechanism of the CDx. |
| CDx | In vitro diagnostic companion diagnostic. |
| Clinical trial assay (Original CDx) | The CDx assay used in the pivotal clinical trial of the associated medicine. |
| Clinical utility | The usefulness of the results obtained from testing with the IVD medical device and the value of the information to the individual being tested or the broader population. |
| Subsequent CDx | An IVD that is different to what was used in the clinical trial but is intended for the same medicine and indication. |
| In-house IVD CDx | An IVD medical device that is:
Refer to guidance on in-house IVDs. |
| Companion testing plan | A companion testing plan is information provided by the sponsor of a medicine or biological, relating to a medicine or biological indication that requires CDx testing, that provides reassurance that Australian patients will be able to safely and effectively use the medicine as they will have access to at least one adequate CDx. |
Page history
Title changed from 'Complying with rules for IVD companion diagnostics (in-vitro diagnostic medical devices)' to 'Understanding regulatory requirements for in vitro diagnostic (IVD) companion diagnostics (CDx)'
Updated guidance to improved clarity on companion diagnostic regulatory requirements including:
- Diagrams to provide an overview of the CDx process
- Introduction of the companion testing plan for medicine sponsors
- Clinical performance requirements for CDx
- Detailed case studies
Title changed from 'IVD companion diagnostics' to 'Complying with rules for IVD companion diagnostics (in-vitro diagnostic medical devices)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Update to reflect the extension in transition arrangements to 26 May 2026 for IVD companion diagnostics.
Reference to 'proposed' removed post approval of regulatory amendments.
Original publication
Title changed from 'Complying with rules for IVD companion diagnostics (in-vitro diagnostic medical devices)' to 'Understanding regulatory requirements for in vitro diagnostic (IVD) companion diagnostics (CDx)'
Updated guidance to improved clarity on companion diagnostic regulatory requirements including:
- Diagrams to provide an overview of the CDx process
- Introduction of the companion testing plan for medicine sponsors
- Clinical performance requirements for CDx
- Detailed case studies
Title changed from 'IVD companion diagnostics' to 'Complying with rules for IVD companion diagnostics (in-vitro diagnostic medical devices)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Update to reflect the extension in transition arrangements to 26 May 2026 for IVD companion diagnostics.
Reference to 'proposed' removed post approval of regulatory amendments.
Original publication