Recalls and market notifications
Understand when and how sponsors must notify the TGA about medical device recalls and market actions under the European Union Medical Device Regulation (EU MDR) transition.
A recall action is a set of market actions that are undertaken, typically via the Uniform Recall Procedure for Therapeutic Goods (URPTG), to resolve a problem with a therapeutic good already supplied in the Australian market for which there are issues, deficiencies or defects in relation to the safety, quality, efficacy (performance) or presentation of the therapeutic good.
There are four distinct recall actions available to sponsors - recall, product defect correction, hazard alert, and product defect alert. Further information on the standard procedures is outlined on the recalls page.
The following illustration provides an overview of how the manufacture of devices and its journey through the supply chain is regarded:
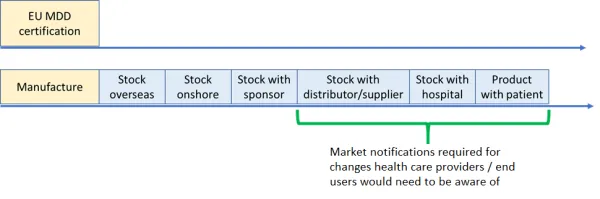
Streamlining of sponsor obligations
In recognition of the scale of the EU MDR transition, and in the interest of minimising regulatory burden on the industry, the TGA is undertaking a risk-based approach in streamlining the type of actions sponsors undertake in meeting their obligations.
If certain criteria are met, sponsors of medical devices transitioning to the EU MDR can:
- notify their customers about changes to their medical devices
- maintain documentation to confirm that the notifications occurred
- provide this documentation to the TGA upon request.
If the changes meet the criteria, sponsors do not need to submit these as separate recall notifications to the TGA Recalls Section.
Eligibility criteria for streamlined market notifications
All of the following 6 criteria will need to be met to qualify for streamlined market notifications:
- The changes being notified only relate to devices transitioning from the EU MDD to EU MDR certification. i.e., the changes are due to a change in regulatory requirements and not because devices currently supplied to the market are unsafe or defective, and
- The devices comply with all Australian regulatory requirements when supplied to the market, and
- There are no deficiencies in safety, quality, performance, or presentation of the devices as currently supplied to the market, and
- The changes being notified are not because of any reported safety related incidents that have resulted in patient or user harm, and
- The changes being notified are not because of any signals arising from adverse event reporting and investigation, and
- The devices were manufactured whilst a conformity assessment certificate was valid.
Sponsors who qualify for streamlined market notifications can either:
- Submit an Online Notification Form to utilise TGA’s EU MDR Transition web publication service to provide market notifications to health care providers and or end users
or
- Notify health care providers and or end users about changes to their devices and maintain documentation to confirm that the notifications had occurred and be able to produce them to the TGA upon request.
Approach to high risk and safety related changes
Sponsors who do not meet the eligibility criteria for streamlined market notification(s) would need to submit new recall notification(s) following the TGA’s established recalls procedure. This includes any high risk and or safety related changes.
The Online Notification Form can also be used to provide market notifications to health care providers and consumers of devices that are not transitioning from the EU MDD to the EU MDR and where supply of the non-transitioning device will cease in Australia.