Understanding the medical device application audit process
This guidance is for sponsors of applications for inclusion of medical devices, including in vitro diagnostic (IVD) devices, in the Australian Register of Therapeutic Goods (ARTG).
Purpose
This guidance is for sponsors of applications for inclusion of medical devices, including in vitro diagnostic (IVD) devices, in the Australian Register of Therapeutic Goods (ARTG).
The application audit process verifies that medical devices meet legislative requirements for inclusion in the ARTG.
This document explains the case management of applications selected for audit and includes both mandatory and non-mandatory application audits.
This document does not apply to device change or variation applications.
Selection for audit
The first step for all medical device applications is preliminary assessment. The TGA must complete preliminary assessment within the statutory timeframe of 20 working days.
Applications that do not pass preliminary assessment will be refused.
If your application passes preliminary assessment, it will either be included on the ARTG or selected for audit.
Any application for a medical device, including IVD medical devices, may be selected for audit using our audit selection criteria.
Note
Some applications must be selected for mandatory audit by law, and an audit fee must be paid.
You do not need to pay an audit fee if your application is selected for non-mandatory audit.
If your application is selected for audit, we will send you a Notice of Selection for Audit and Requirement to Provide Further Information (s41FH) according to the Therapeutic Goods Act 1989 (‘the Act’).
This will explain the legal basis of selecting your application for mandatory audit or why we selected your application based on the selection criteria for non-mandatory audit.
We will let you know what extra information we need for the audit, and what documents to provide.
For mandatory audits, we will also invoice the audit fee that you will need to pay for the application to proceed.
We cannot start the audit until the fees have been paid.
During the audit process, we will review any matters raised in preliminary assessment in more detail.
We will also ask for evidence of compliance with legislative requirements including the Essential Principles, and we may ask for clarification of information already submitted with your initial application.
Case management
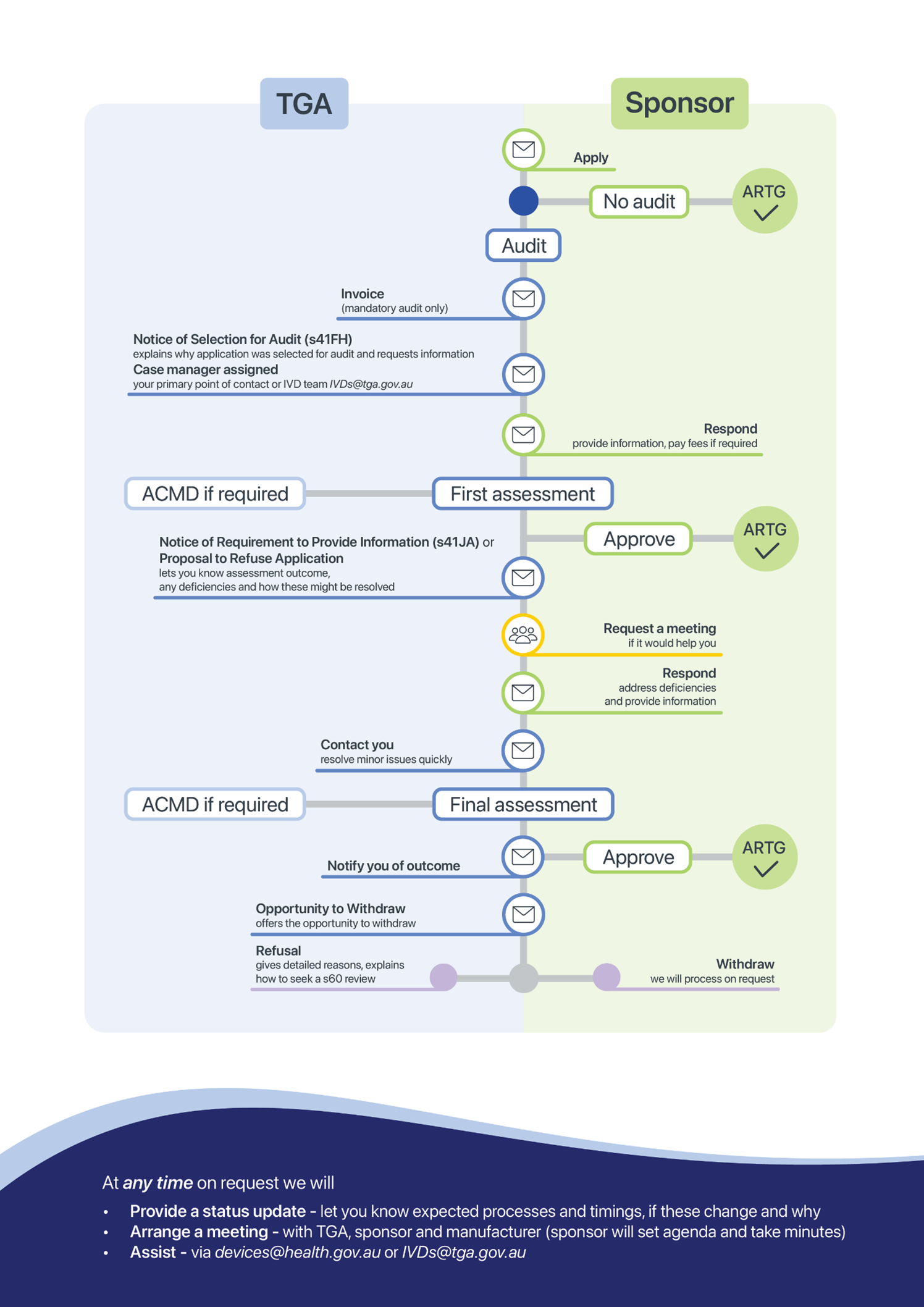
Figure 1 gives an overview of the case management process for all medical device application audits.
The figure shows two rounds of assessment. More rounds of assessment will be considered in some cases. We will always give you a chance to respond to issues before we decide your application.
You can request a meeting with the TGA at any time during the audit process. We will provide you with our best contact details on every communication we send you.
We understand you need us to communicate well during the audit process. We are improving the clarity of our communication and our processes.
We will aim to provide timely and regular updates on your application status, expected timeframe, and days that have elapsed.
We will also phone or email you informally to resolve simple matters.
Figure 1. Application audit case management process (including IVDs)
{kind=link}
This flowchart illustrates the end-to-end process for managing applications to the ARTG via the TGA, specifically when audits are involved.
It is structured into two primary roles: Sponsor and TGA, with directional arrows indicating the flow of actions and decisions.
Process overview
- Application initiation
The process begins with the Sponsor submitting an application to the TGA. - Audit determination
- If no audit is required, the application proceeds directly to ARTG approval.
- If an audit is required, the TGA issues an invoice for a mandatory audit.
- Audit notification
The Sponsor receives a Notice of Selection for Audit (s41FH). This includes a request for information and the assignment of a case manager as the primary contact. - Sponsor response
The Sponsor must respond by providing the requested information and paying any applicable fees. - First assessment by TGA
- If necessary, input from the Advisory Committee on Medical Devices (ACMD) is sought.
- If deficiencies are identified, the TGA may issue a Notice of Requirement to Provide Information (s41JA) or a Proposal to Refuse Application.
- Approval or further action
- If the application is satisfactory, it proceeds to ARTG approval.
- If issues remain, either party may request a meeting to discuss and resolve them.
- Sponsor follow-up
The Sponsor addresses deficiencies and submits additional information. - TGA contact
The TGA may reach out to the Sponsor if further issues arise during this stage. - Final assessment
- Further ACMD input may be required.
- The TGA then notifies the Sponsor of the outcome, which may be approval or refusal.
- Outcome options
- Opportunity to Withdraw: Sponsors may withdraw their application at any point.
- Refusal: Applications may be refused under s60 review.
- Approval: Successful applications are added to the ARTG.
Support and transparency
At any stage, upon request, the TGA will:
- Provide a status update detailing expected timelines and any changes.
- Arrange a meeting involving the TGA, Sponsor, and Manufacturer (Sponsor sets the agenda and takes minutes).
- Assist via email at:
Non-IVD applications
If your medical device application is selected for audit, you will be assigned a case manager who will contact you.
We will let you know if your case manager changes.
If we don’t need you to provide information for the audit, we will assign a case manager straight away.
Otherwise, your case manager will be assigned when we receive your response to the Notice of Selection for Audit and Requirement to Provide Further Information (s41FH).
We understand it would help sponsors to be assigned a case manager earlier, and we are improving our system to allow this.
Your case manager will:
- be your primary contact during the audit process
- monitor, track and communicate the progress of your audit over time
- coordinate assessments by the TGA’s specialist assessment teams or external experts
- communicate and discuss any deficiencies and information requests from assessment areas
- facilitate meetings between TGA assessors, you and your manufacturer when required
- if required, coordinate with the Advisory Committee on Medical Devices (ACMD) for advice.
IVD applications
If your IVD application is selected for audit, the IVD application management team will contact you through the IVD inbox.
We understand it would help sponsors to be assigned a dedicated case manager for IVDs, and we are working towards this model.
If we don’t need you to provide information for the audit, the application will move to assessment.
Otherwise, the application will move to assessment when we receive your response to Notice of Selection for Audit and Requirement to Provide Further Information (s41FH) and the audit fees (where applicable).
Apart from having a dedicated case manager, the IVD application management team provides the same case management as we provide for non-IVD medical device applications.
Types of audit
We have two types of audits: Level 1 and Level 2 audits.
On rare occasions, Level 1 audits may become Level 2 because specialist assessment is needed.
Level 2 audits may become Level 1 if we find that specialist assessment is considered no longer needed.
Your case manager will let you know if the type of audit changes.
Level 1 audits
Level 1 audits do not need specialist assessment. Level 1 audits verify compliance with matters such as patient implant cards, patient information leaflets, labelling, instructions for use, classification, or conformity assessment documentary requirements.
Level 2 audits
Level 2 audits are more complex and usually require specialist assessment such as clinical, software, engineering, biomaterials, or microbiology.All IVD audits are level 2 audits.
- Non-mandatory IVD audits assess the specified audit selection criteria
- Mandatory IVD audits assess the manufacturer’s technical file for the device
- IVD audits require specialist assessment by our IVD assessors and may also require clinical or software assessment.
For more details on IVD audits see Preparing a technical file review for IVD medical devices.
Timeframes
We monitor our processing time for each application in working days in the Australian Capital Territory.
Our timing starts when the application is submitted via the TGA Business Services Portal, and the application fee has been paid, if necessary.
We aim to complete application audits within the following targets for processing time:
- Level 1 audits: 50 working days
- Non-mandatory Level 2 and IVD audits: 150 working days
- Mandatory Level 2 and IVD audits: 180 working days
Level 1 audit timeframes do not apply for IVD medical devices as all IVD audits are Level 2.
The timeframes include the statutory preliminary assessment period of up to 20 working days, waiting periods for specialist assessment at the TGA, and any meetings with us that you request.
The timeframes do not include any time when we are waiting for you to respond to a notice, letter or request for information or pay any assessment fees.
These target timeframes are based on actual processing times.
The targets supersede the historical arbitrary 60 working day target for application audits that was not consistently met.
We are committed to improving these timeframes.
We will be improving our processes and will review our progress in 12 months.
Non-IVD assessment timeframes
For non-IVD Level 2 application audits, we expect the first round of specialist assessment to be completed in 100 working days.
If this is not possible, your case manager will let you know and explain the revised timeframe.
IVD assessment timeframes
We will let you know when we start the first and second rounds of assessment and let you know the estimated timeframe for completion for each round. If this changes, we will let you know and explain the revised timeframe.
We aim to complete all IVD application audits in the target timeframes.
Advisory Committee on Medical Devices
At any stage during the audit process, we may seek specialist advice from the Advisory Committee on Medical Devices (ACMD) about your application.
We will notify you if this happens and let you know the questions we will ask the committee.
Before the ACMD meeting, you can submit comments in response to the questions we will ask the committee, although this is optional. If you choose to respond, do so within two weeks.
We will provide your response to the committee to consider when giving us its advice.
We will let you know the ACMD’s advice when the meeting minutes are ratified.
Assessment outcomes
Your case manager will review the specialist assessments and any identified deficiencies.
You can ask your case manager or the IVD team to clarify issues or to arrange follow-up meetings at any time.
Approval
If no deficiencies are found in the first specialist assessment, the application will be approved.
Deficiencies
If deficiencies are identified, you will receive a Notice of Requirement to Provide Information (s41JA) explaining the deficiencies and ask you to clarify or provide more information to address the deficiencies.
Significant deficiencies
If we see significant non-compliance, we will send you a Proposal to Refuse Application.
This gives reasons for our views, including details of the significant deficiencies.
This is not a final decision. We encourage you to respond to this letter.
Response
After receiving a Notice of Requirement to Provide Information (s41JA) or a Proposal to Refuse Application, you can address the deficiencies in writing and provide further information.
Depending on the nature of the deficiencies you will have between 20-40 business days to respond.
If the deficiencies are complex or substantial, you can ask for more time.
When you respond, we will assess whether the deficiencies have been clarified or addressed. We might ask for further information.
Refusal or withdrawal
If all the deficiencies are not overcome, we will advise you by email that we are planning to refuse your application.
You will have two weeks to withdraw your application before we proceed to refusal. You can ask us if you need more time.
Some sponsors may decide to withdraw the application and so avoid refusal. If an application is withdrawn or refused, we cannot refund application or assessment fees.
You cannot seek a review if you withdraw your application.
We will explain our reasons in the written notice of the decision to refuse your application.
We will also explain how you can seek a review of the decision under s60 of the Act. There is no additional fee associated with a review under s60.
Page history
Original publication.
Original publication.