Pharmacovigilance responsibilities of medicine sponsors
Australian recommendations and requirements
Introduction
Pharmacovigilance is defined by the World Health Organization as the science and activities related to detecting, assessing, understanding and preventing adverse effects and other medicine-related problems. The Therapeutic Goods Administration (TGA) collects and evaluates information related to the benefit-risk balance of medicines in Australia to monitor their safety and, where necessary, take appropriate action.
This guidance sets out the pharmacovigilance responsibilities of sponsors of medicines included on the Australian Register of Therapeutic Goods (ARTG) and regulated by the TGA. It outlines the mandatory reporting requirements and offers recommendations on pharmacovigilance best practice.
In this guidance we use ‘must’ or ‘required’ to describe something you are legally obliged to do. We use ‘should’ to recommend an action that will assist you to meet your legal requirements. We refer to the TGA as ‘we’ or ‘us’, and to sponsors as ‘you’.
Scope
This guidance:
- applies to all sponsors who have medicines registered or listed on the ARTG
- describes your pharmacovigilance reporting and record-keeping requirements
- offers recommendations on the monitoring, collection and management of safety data to help you achieve best practice pharmacovigilance
- outlines the legal basis for pharmacovigilance requirements associated with medicines on the ARTG.
This guidance is mostly consistent with the European Medicines Agency’s EMA Guideline on Good Pharmacovigilance Practices (GVP) Module VI—Management and reporting of adverse reactions to medicines. Some requirements and recommendations, however, are specific to Australia.
Responsibilities
You must meet your pharmacovigilance reporting responsibilities for all the medicines you have registered or listed on the ARTG. This is regardless of their Australian marketing status—that is, whether they are currently available for purchase, withdrawn from sale or otherwise supplied (e.g. in a company-sponsored post-registration study).
Unapproved medicines used in clinical trials or supplied under the Special Access Scheme or Authorised Prescriber Scheme are subject to separate reporting requirements and are not covered by these guidelines.
You, as a sponsor of medicines approved for supply in Australia, are legally responsible for meeting pharmacovigilance reporting requirements for your medicine.
You must:
- let us know who your Australian pharmacovigilance contact person is
- submit any serious adverse reaction reports to us
- notify us of any safety issues you identify
- keep records pertaining to the reporting requirements and safety for your medicine (under Subsection 28(5)(ca) of the Therapeutic Goods Act 1989 (the Act))
- answer any request from us for additional information fully and within the specified timeframe (under Subsection 31(1) of the Act).
This includes situations where:
- separate sponsors market two or more separately registered or listed medicines that are considered identical in all respects apart from their trade names
- sponsors share or outsource marketing or distribution arrangements
- pharmacovigilance activities are contracted to external organisations.
We require you to have an effective pharmacovigilance system in place in order to:
- collect and collate safety information pertaining to your medicine
- monitor and take responsibility for the safety of your medicine
- meet legislative requirements for reporting serious adverse reactions and safety issues
- identify any changes to the benefit-risk balance of your medicine
- take appropriate action in a timely manner when necessary
- update product labels and product information (PI) documents with new safety information in a timely way.
Related information and guidance
Questions relating to this guidance document may be addressed to Pharmacovigilance.Enquiries@health.gov.au
Your regulatory reporting requirements
Your pharmacovigilance reporting requirements (i.e. what, how and when you MUST report) are summarised in the table below.
| Report type | How to report | Reporting timeframe |
|---|---|---|
| Australian pharmacovigilance contact person | Submit via TGA Business Services | ≤15 calendar days of first ARTG entry or of any updates to details. |
| Significant safety issues | Submit a notification using the online Medicine safety issues – Electronic notification form | ≤72 hours of Australian sponsor awareness and of any follow-up information. |
| Other safety issues | ≤30 calendar days of Australian sponsor awareness and of any follow-up information. | |
| Serious adverse reaction reports that occurred in Australia | Submit a CIOMS form via email to: adr.reports@health.gov.au or submit report online using the Adverse Event Management System (AEMS): TGA Business Services, or submit via E2B reporting | ≤15 calendar days of Australian sponsor awareness and of any follow-up information. |
| Quality defect issues including adulterated and counterfeit products | Report as per above depending on report type and also refer to the Procedure for recalls, product alerts and product corrections to determine the appropriate market action or recall | In accordance with the timeframe for safety issues or serious adverse reactions, as applicable. |
| Non-serious adverse reaction reports and overseas adverse reaction reports | Report as per above depending on report type and also refer to the Procedure for recalls, product alerts and product corrections to determine the appropriate recall or market action. | As specified by the TGA PSUR reporting requirements or specific request. |
Note
Timeframes are in relation to Day 0.
Day 0 for each report type is defined in the relevant section of this document.
What, when and how to report
Australian pharmacovigilance contact person
Nominate a person in Australia to be responsible for fulfilling your reporting requirements for all medicines you sponsor in Australia. This person will be the primary direct contact for all pharmacovigilance correspondence between you and the TGA. You may nominate more than one person.
You must:
- provide us with the name and contact details of the Australian pharmacovigilance contact person (A-PVCP) within 15 calendar days of your first medicine’s entry on the ARTG (Day 0).
- notify us of the updated name and contact details within 15 calendar days of any changes to the A-PVCP or their details. Day 0 is considered to be the date of the change of the person, or the existing A-PVCP’s details (e.g. email address).
The A-PVCP must be nominated, and/or their details updated, through the TGA Business Services (TBS) electronic portal. For further assistance, please contact the TBS helpdesk.
You should regularly check your TBS profile to ensure the A-PVCP’s details remain correct.
The A-PVCP must reside in Australia and should have a sound understanding of the Australian pharmacovigilance reporting requirements. Please note that the A-PVCP may be different to the qualified person responsible for pharmacovigilance in Australia (QPPVA) although ideally, they are the same person.
Serious adverse reactions
Pharmacovigilance distinguishes between adverse events and adverse reactions.
Adverse events
An adverse event is any untoward medical occurrence in a patient, consumer or clinical investigation subject administered a medicine, which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign (e.g. an abnormal laboratory finding), symptom or disease temporally associated with the use of a medicine, whether or not it is considered related to the medicine.
Adverse reactions
An adverse reaction is a noxious and unintended response to a medicine. The phrase ‘response to a medicine’ means there is at least a reasonable possibility that the medicine caused the adverse event. A reaction, in contrast to an event, is characterised by the fact that a sponsor, investigator or reporter suspects there is a causal relationship between the medicine and the occurrence.
This guidance describes the reporting requirements that apply to adverse reactions (as opposed to adverse events) identified from all sources, including adverse reactions that arise from:
- the use of a medicine within the terms of approved indications as per the PI document or ARTG
- the use of a medicine outside the terms of the approved indications—including overdose, abuse, misuse, off-label use and medication errors
- occupational exposure.
Note
Synonyms of adverse reaction include adverse drug reaction (ADR), suspected adverse (drug) reaction, adverse effect, and undesirable effect.
For regulatory reporting purposes, if an adverse event is spontaneously reported, even if the causal relationship is unknown or unstated, it meets the definition of an adverse reaction. Therefore, all spontaneous reports notified by healthcare professionals or consumers are considered suspected adverse reactions, since they convey the suspicions of the primary sources, unless the reporter specifically states they believe the events to be unrelated or that a causal relationship can be excluded.
You should make reasonable attempts to obtain additional information so you can assess causality.
Expected and unexpected adverse reactions
An expected adverse reaction is an adverse reaction known to be associated with the use of the medicine, as reflected in the PI document or label warning statement.
An unexpected adverse reaction is an adverse reaction in which its nature (that is, its specificity or outcome), severity or frequency is either not identified, or not consistent with the term or description used, in the PI document or label warning statement.
Regulatory reporting requirements apply to both expected and unexpected adverse reactions.
Serious adverse reactions
A serious adverse reaction is any medical occurrence that, in relation to a medicine, at any dose:
- results in death
- is life-threatening
- results in inpatient hospitalisation or prolonged hospitalisation
- results in persistent or significant disability or incapacity
- is associated with a congenital anomaly or birth defect (see Reports in pregnancy and breastfeeding)
- is a medically important event or reaction.
Non-serious adverse reactions
A non-serious adverse reaction is an adverse reaction that does not meet the definition of a serious adverse reaction.
What you must report
You must report any of the following of which you are aware:
- expected and unexpected serious adverse reactions associated with the use of your medicine that occurred in Australia, including those reported in the published international or local scientific or medical literature
- all clinically and medically relevant follow-up information related to serious adverse reaction reports occurring in Australia.
All serious adverse reaction reports should be validated (see Validating adverse reaction reports) and followed up as necessary (see Following up adverse reaction reports).
You must report all serious adverse reactions that occur in Australia as soon as possible and no later than 15 calendar days from receipt. The clock for serious adverse reaction reporting starts (as Day 0) on the day that the four minimum data elements in relation to the adverse reaction report are received by any of your personnel—including sales representatives and contractors.
Where you have entered into a relationship with a second company, the timeframe for regulatory submission should be no longer than 15 calendar days from first receipt by personnel of either company. Explicit procedures and detailed agreements should exist between you and the second company to facilitate achievement of this objective.
The seriousness of the report is linked to the seriousness of the reported adverse reaction. It is important that seriousness assessments are an independent process to medical evaluation, causality and validity of the case and are based on the adverse reaction alone. Where outcomes or treatment information (e.g. hospitalisation) is not available, a conservative approach should always be taken.
You do not need to report serious adverse events, including from spontaneous reports, where:
- there is no plausible temporal or causal association with the medicine
- the reporter specifically states they believe the events to be unrelated or that a causal relationship can be excluded, and you agree with this assessment.
For the individual adverse reaction reports not required to be reported to us (such as non-serious adverse reaction reports, serious adverse reaction reports from overseas and invalid adverse reaction reports), you:
- Must keep records of these adverse reactions and provide these to us if we request them, in the specified format and timeframe
- Must include these adverse reactions as a summary tabulation in a PSUR, if one is required
- Must report adverse reactions that have a major impact on the benefit-risk balance or overall safety profile of the medicine as a significant safety issue.
Serious adverse reactions occurring overseas in Australian patients using medicines supplied from Australia, for example while the patient is travelling on holiday, should be treated as an Australian case and must be reported to us.
Adverse reactions from literature
You must report all serious adverse reaction cases occurring in Australia for your medicine that are identified through screening the worldwide literature as soon as possible and no later than 15 calendar days from receipt. Day 0 is when you become aware of a publication containing the four minimum data elements.
We prefer that you include a copy of the relevant published article (in English or an English summary/translation) when you make the initial adverse reaction report. However, if the article is not available at this time you should provide it to us within 15 calendar days of submitting your report. If you have difficulty meeting this timeframe, notify us in writing prior to the 15-day period ending. For serious adverse reaction reports from the literature submitted via E2B, inclusion of the publication reference in Vancouver style within the E2B report is sufficient.
You may use the services of an external party to conduct searches of the published scientific and medical literature; however, you remain responsible for the performance of the search and subsequent reporting. Day 0 is when anyone, either of the sponsor or the contractual partner (whoever is the earliest), becomes aware of a publication containing the minimum information. The transfer of a pharmacovigilance task or function should be detailed in a contract between you and the service provider, to ensure that published literature adverse reaction cases are reported as required within the correct timeframes. Where a third party provides a review or a collated report from the published scientific and medical literature, Day 0 is the date the search was run (provided the minimum criteria are available in the abstract), and not the date the information was supplied to your company.
Validating adverse reaction reports
Only reports of serious adverse reactions that include the minimum four data elements are considered valid and required to be reported.
The minimum 4 data elements are:
- one or more identifiable reporter(s) (the primary source)—such as their qualification, name, initials, address or contact details (for follow-up)
- an identifiable patient—such as their initials, gender, patient identification number, date of birth, age or age group
- one or more suspected medicine(s)
- one or more suspected reaction(s).
Therefore, all reports of suspected serious adverse reactions should be validated before you submit your report to the TGA. If a report cannot be validated, it should still be retained and recorded in your pharmacovigilance system.
It is important to identify the patient and the reporter to avoid case duplication and fraudulent reporting and allow cases to be followed up as required. In this context, identifiable means you can verify the existence of a real patient or a reporter. If you believe that there is a real patient involved (without any identifiers), it is considered sufficient for reporting. If the reporter does not wish to provide contact details, the adverse reaction is still considered valid if you can confirm the case directly with the reporter at the point of the initial report. If the report is second-hand, make every reasonable effort to verify the existence of an identifiable patient and reporter. All parties providing case information or approached for case information should be identifiable, not only the initial reporter.
A serious adverse reaction report that lacks any of the above four items is invalid and does not qualify for reporting. Invalid reports also include those where:
- the primary source has explicitly stated that a causal relationship between the medicine and event can be excluded, and you agree
- the type of adverse reaction is unspecified
- only an outcome or consequence, such as hospitalisation or death, has been reported, with no further information provided on clinical circumstance to consider it a suspected adverse reaction.
A report that is determined to be invalid may still provide pertinent safety information.
Assess reports of adverse reactions on a case-by-case basis. Use your clinical judgement to determine whether it is a valid serious adverse reaction or could constitute a safety issue and must be reported—for instance, you should report unexpected sudden deaths where the reporter considers it related to the suspect medicine or clusters of drug-event pairs that may indicate a safety signal.
You should exercise due diligence in following up invalid adverse reaction reports to collect missing data elements. Nevertheless, all reports of adverse reactions regardless of their validity should be recorded in your pharmacovigilance system for use in ongoing safety analysis activities. If you do not report a serious adverse reaction to the TGA, you should document the reason for this.
Following up adverse reaction reports
Initial adverse reaction reports received by the sponsor that do not include the minimum four data elements should be followed up as necessary to obtain the missing information.
You should also follow up adverse reaction cases to obtain detailed supplementary information significant to the clinical evaluation of the cases. This is particularly important for:
- monitored events of special interest (e.g. identified or potential risks and missing information in the Risk Management Plan (RMP))
- prospective reports of pregnancy (See Reports of exposure during pregnancy and breastfeeding)
- cases notifying the death of a patient
- cases reporting new risks or changes in the known risks of your medicine.
You should document your attempts to obtain follow-up information.
Tailor your follow-up to optimise the collection of missing information, in a way that encourages the primary source to submit relevant new information.
For consumer reports of serious adverse reactions, if the information is incomplete, you should attempt to obtain the consumer’s consent to contact their healthcare professional to confirm the report and add medical information as required. You cannot downgrade a valid report of an adverse reaction from a consumer if their healthcare professional disagrees with their suspicion (in terms of relatedness and seriousness). In this situation, you should include the opinions of both the healthcare professional and the consumer in your report to us.
The reporting time clock restarts when you receive additional clinical or medically relevant information for a previously reported serious adverse reaction.
Only significant follow-up information MUST be reported to the TGA, for example, new information about an adverse reaction initially classified as non-serious indicates the case should be re-classified (i.e. from non-serious to serious).
You must report this information as soon as possible and no later than 15 calendar days after you receive the additional information.
Key data elements for adverse reaction reports
We recommend that you try to collect and include as many of the below key data elements about the adverse reaction as possible in the report so it can be assessed. Some information might not be relevant, depending on the circumstances.
Reporter details
- Name
- Mailing address and/or email address
- Telephone number
- Reporter type (e.g. consumer, healthcare professional etc.)
- Profession (specialty e.g. physician, pharmacist, other healthcare professional; lawyer, consumer or other non-healthcare professional).
Patient details
Do not report the patient’s name to us. We record only the information necessary for us to perform our regulatory function.
- Initials
- Any other relevant identifier such as patient number
- Gender
- Age, age category (e.g. adolescent, adult or elderly) or date of birth
- Concurrent conditions
- The patient’s medical history including relevant past medicine history
- Relevant family history
- Weight
- Height
- Ethnicity
- Aboriginal and/or Torres Strait Islander origin status
- For reports about maternal/paternal or foetal exposure (see Reports of exposure during pregnancy and breastfeeding):
- the gestation period at time of exposure
- information about the parent (e.g. their identity, age or date of birth, date of last menstrual period, weight, height, gender, relevant medical history and concurrent conditions, relevant past medicine history).
Details of the suspected medicine(s)
- Brand name as reported
- International Non-Proprietary Name (INN) or Australian Approved Name (AAN)
- The AUST R or AUST L number on the label
- Active ingredient batch or lot number
- Indication(s) for which suspect medicine was taken, prescribed or tested
- Dosage form and strength
- Daily dose in units (e.g. mg, mL, mg/kg) and regimen
- Administration route
- Administration site
- For reports about maternal/paternal or foetal exposure, route of administration to parent (see Reports of exposure during pregnancy and breastfeeding)
- Starting date and time
- Stopping date and time or duration of treatment
- Any changes to medicine administration (e.g. medicine withdrawn, dose reduced, dose increased, dose not changed)
- Any additional relevant information on the medicine
- If the reaction is suspected to be the result of an interaction with alcohol, food or another medicine, the names and active ingredients of the suspected interacting medicines or substances.
Other treatment(s)
Provide the same information as for suspected medicines for:
- concomitant medicines including non-prescription, over-the-counter medicines, herbal remedies, dietary supplements, complementary and alternative therapies etc.
- relevant medical devices.
Details of the adverse reaction(s)
- A full description of the reaction(s), including body site and severity
- The primary source’s description of the reaction, verbatim
- A description of the reaction using the lowest level terms in the Medical Dictionary for Regulatory Activities (MedDRA) terminology
- Why the report is considered serious
- A description of the signs and symptoms
- A specific diagnosis of the reaction
- The date and time of the reaction’s onset
- When the reaction ceased, or its duration
- The interval between when the suspect medicine was administered and the reaction
- Dechallenge and rechallenge information
- Results and laboratory data from relevant tests
- Location where the reaction occurred (e.g. hospital, outpatient clinic, home or nursing home)
- The outcome of the reaction when last observed (e.g. recovered/resolved, recovering/resolving, not recovered/unresolved or recovered/resolved with sequelae—describe the sequelae)
- If relevant, date of death
- Whether an autopsy was performed and, if so, any relevant autopsy or post-mortem findings, including coroner’s report
- Cause of death for a fatal outcome
- Relatedness of medicine to reaction(s)/event(s)
- Assessment of reaction—source of assessment (e.g. initial reporter, investigator, regulatory agency, company), method of assessment (e.g., global introspection, algorithm, Bayesian calculation) and result
- Case narrative including clinical course, therapeutic measures, outcome and additional relevant information
- Sponsor’s comments (e.g. diagnosis/syndrome and/or classification of reaction/event)
- Whether the case was medically confirmed, including medical documentations (e.g. laboratory or other test data) provided by either a consumer confirming the adverse reaction occurred, or an identifiable healthcare professional)
- Document the whole report as a medically confirmed spontaneous report if:
- a consumer initially reports more than one reaction and at least one reaction was medically confirmed
- a medically qualified patient, friend, relative of the patient or carer submits a report.
Administrative and sponsor details
- Source of report (e.g. spontaneous, epidemiological study, patient survey, literature, etc.)
- Date the event report was first received by sponsor
- Country in which the event occurred
- Type (initial or follow-up) and sequence (first, second, etc.) of case information reported to authorities
- Name and address of sponsor
- Name, address, email address and telephone number of pharmacovigilance contact person at the sponsor’s Australian address
- Company/manufacturer's identification number for the case (the same number should be used for the initial and follow-up reports on the same case)
- The ADR identification number (if known) of possible duplicate reports initially submitted to the TGA by a consumer, healthcare professional or other primary source.
Content of an adverse reaction report
Ensure:
- your report complies with Part A of the General dossier requirements
- any computer-generated forms are legible, contain the appropriate content and follow the approved layout
- you do not reduce or condense the report.
You must provide the original words the reporter used to describe the adverse reaction. We prefer that you also include the appropriate lowest level terms from MedDRA in your adverse reaction report.
If you have sufficient information from the primary source to prepare a concise clinical summary of the individual case, provide a case narrative consistent with the data in other parts of the report. You should present the information in a logical chronological sequence as the patient experienced it, including:
- the clinical course
- any therapeutic measures taken
- the outcome
- relevant follow-up information.
You should summarise any relevant autopsy or post-mortem findings. You may include your opinion on whether there is a causal association between the suspected medicine(s) and reaction(s), and details of the criteria you used to make this assessment.
Where you cannot obtain consent to disclose the personal details of the patient or reporter, or to contact the treating doctor for medical confirmation of consumer reports, indicate this in your report as a sponsor comment.
For follow-up reports, you should clearly highlight what follow-up information has been provided and reference the unique TGA adverse event record number designated to the initial report (stated in your acknowledgment letter from the TGA).
If the report is likely to be a duplicate—for instance, if you are aware the reporter has reported the adverse reaction to the TGA or it involves multiple suspected medicines—let us know this in your report. You should provide all available details to help us identify the duplicate including the TGA adverse event record number allocated to the initial report, if known.
If the reaction is suspected to be the result of an interaction with alcohol, food or another medicine, you should state this clearly in the report and list the suspected interacting products or substances. For combination medicines that contain more than one active ingredient, list each active ingredient. If the primary source suspects a possible causal role of one of the ingredients in the medicine, provide this information in the report.
Related information and guidance
Safety issues
Your requirement to report safety issues relating to your ARTG-listed or registered medicines is underpinned by legislation in Sections 29A and 29AA of the Therapeutic Goods Act 1989. Refer to the section on Pharmacovigilance and the law in this document for more information.
We use the information that you report to us about safety issues to take appropriate action. This may include providing further safety information to the public, requesting updates to PI documents, imposing additional risk management interventions or pharmacovigilance activities, or (rarely) removing a medicine from the market.
Where you have contractual arrangements with a third party (external) person, other sponsors or external organisations, your pharmacovigilance contract or agreement should outline roles, responsibilities and timelines to ensure you can comply with your reporting obligations for safety issues.
If you determine after appropriate assessment that a safety issue does not require reporting to us, you should document a justification for this decision. We may ask you to provide this documentation at any time.
A safety issue reporting decision tree is included as an appendix to this document. If you are still in doubt about whether to report a safety issue, contact the PB Signal Investigation Coordinator (si.coordinator@health.gov.au) for advice.
Significant safety issues
A significant safety issue (SSI) is a safety issue relating to your ARTG-listed or registered medicine that requires the urgent attention of the TGA as it is likely to warrant prompt regulatory action. It is significant because of the seriousness and potential major impact to the benefit-risk balance of the medicine and/or public health.
Type of SSIs include:
- the following major safety-related actions by comparable overseas regulators (CORs):
- the withdrawal or suspension of the medicine’s availability
- the modification or removal of an indication (for safety reasons), or the addition of a boxed warning or contraindication to the PI document or label.
- major safety issues identified in the context of an ongoing or newly completed non-clinical study, post-registration study or clinical trial (e.g. an unexpected increase in the rate of fatal or life-threatening adverse events)
- major safety issues identified through spontaneous reporting or published literature, which may lead to the addition of a contraindication, restriction of the use of a medicine, or its withdrawal from the market.
- serious quality defect issues that may lead to an immediate risk to public health.
Professional judgement should be used to determine whether a safety issue is significant. Each safety issue should be assessed on a case-by-case basis and evaluated to determine whether it has a major impact on the medicine’s benefit-risk balance and/or implications for public health.
Significant safety issues must be reported as soon as possible and no later than 72 hours from awareness. The clock for reporting (Day 0) begins as soon as personnel of the Australian sponsor (including any third parties, vendors or partners that have been delegated pharmacovigilance responsibilities) becomes aware of a safety issue from any source that may meet the definition of an SSI.
We recognise that safety information may be received and processed by your global counterparts before it is disseminated to the local Australian sponsor. In this situation, we expect you to have clearly documented internal procedures in place to ensure expedited communication of SSIs from global personnel to your relevant Australian personnel for reporting. Your procedures should ensure that SSIs will be communicated from global to Australian personnel in no more than 3 calendar days from global awareness, and more rapidly for the most serious safety issues. You must keep records of communications including dates when global and local personnel were notified of SSIs and reasons for any delays in communication.
The reporting clock restarts when you receive additional clinical or medically relevant information related to a previously reported SSI.
Other safety issues
Other safety issues (OSIs) are safety issues that may require action by the TGA but do not need to be actioned urgently. These issues are unlikely to significantly alter the benefit-risk balance of the medicine.
Types of OSIs include:
- safety-related changes, requested by CORs, to the PI documents or labels (that do not meet the definition of an SSI) that have been internally assessed, irrespective of whether you agree with the conclusions or recommendations of the regulator
- safety issues from any other source that have been internally assessed and confirmed with subsequent risk mitigation strategies determined (that do not meet the definition of an SSI).
Your internal assessment should include a description of the safety issue, the source and any available evidence, your assessment of the risk and potential impact of the safety issue, and any action you propose to take in Australia (e.g. PI update) – or justification for no further action.
OSIs must be reported to the TGA within 30 calendar days. Day 0 is the day that any personnel of your Australian sponsor (including any third parties, vendors or partners that have been delegated pharmacovigilance responsibilities) are made aware of an assessed safety issue.
We recognise that safety information may be received and processed by your global counterparts before it is disseminated to the local Australian sponsor. In this situation, we expect you to have clearly documented internal procedures in place to ensure timely communication of OSIs from global personnel to your relevant Australian personnel for reporting. You MUST keep records of your communications including dates when global and local personnel were notified of OSIs and reasons for any delays in communication. Substantial or inappropriate delays between the global and Australian notification of OSIs may be considered non-compliance with regulatory reporting timeframes.
The reporting clock restarts when you receive additional clinical or medically relevant information related to a previously reported OSI.
How to report a safety issue
All safety issues should be reported to the TGA by submitting a notification using the online Medicine safety issues – Electronic notification form.
To assist us in evaluating each safety issue you must provide us with any additional information we request.
Where a safety issue will result in urgent changes to the Australian PI document and you are liaising with a clinical delegate regarding the issue, please copy the Signal Investigation Coordinator in your communications to the applicable TGA clinical delegate.
Notification of COR-identified safety issues by sponsors of generic medicines
If you are the sponsor of a generic medicine, your obligation to report a safety issue from a COR action or request is fulfilled with the submission of an application to align the PI document of your generic product with the Australian innovator PI document, within one month of the date of approval of the safety-related update to the Australian innovator PI document.
Where there is no Australian innovator product to your generic product, or if the Australian innovator product is withdrawn from the ARTG, the obligation to identify and report safety issues from COR actions or requests lies with you, as per the timeframes outlined in the Significant safety issues and Other safety issues sections above.
Safety issues identified by the sponsor of a generic product through any other source (e.g. internal ongoing analysis of the benefit-risk balance of the medicine) must be reported to the TGA as per the requirements outlined in the Significant safety issues and Other safety issues sections above.
Reporting requirements for special situations
Reports from post-registration studies
You must report all serious suspected adverse reactions, of which you are aware, relating to the studied (or supplied) medicine, that occur in post-registration studies undertaken in Australia, in accordance with the reporting timeframes for serious adverse reactions.
You must report any major safety issues identified during post-registration studies which have a potential major impact to the benefit–risk balance of the medicine as a significant safety issue.
This includes post-registration studies for your medicine which:
- use multiple or combined medicines in the trial.
- uses blinding. You are only required to report serious adverse reactions if (or when) the blinding is broken.
- are conducted by a contracted third party (i.e. company-sponsored study). You should have an agreement in place to ensure you can fulfil your reporting requirements.
The above reporting requirements apply only to post-registration studies where your medicine is being used in line with the PI or label indications; for other situations, follow the clinical trials reporting guidelines.
You should have mechanisms in place to:
- collect full and comprehensive information on any adverse event(s)
- follow up reports and seek the primary source’s opinion on causality
- evaluate information and assess individual cases in a timely manner
- report valid adverse reactions related to the studies (or supplied) medicine in accordance with the reporting requirements.
You do not need to routinely submit individual adverse reaction reports for:
- non-serious adverse reactions
- adverse events not suspected to be related to the medicine by the investigator
- adverse reactions that occurred overseas during post-registration studies.
However, you must retain records of these non-reportable cases to be considered in ongoing global analysis of the benefit–risk balance of the medicine and provide these to us if requested.
In instances where the post-registration study is conducted or initiated by an investigator independent of the sponsor of the medicine, the responsibility for reporting adverse reactions to us rests with the investigator. However, if you are aware of the study, you should request to be notified by the investigator of serious adverse reactions that occur in the study. Where you become aware of such adverse reactions, you must ensure that the adverse reactions are reported to us as required.
Reports from other post-marketing initiatives
If you are undertaking in other post-marketing initiatives that include collecting information related to your medicine, you may receive reports of adverse events.
Such initiatives might include:
- patient support, product familiarisation and disease management programs
- programs concerning sponsor supply of medicine
- surveys of patients or healthcare providers
- collecting information on efficacy or patient compliance
- market research programs; and
- voluntary patient registries.
You should have a system in place for collecting and evaluating comprehensive case information so you can determine whether the adverse events are potentially related to the studied (or supplied) medicine. Refer to the section on solicited reports for further information.
You must report suspected serious adverse reactions received from post-marketing initiative reports in accordance with the reporting requirements for serious adverse reactions.
Reports of exposure during pregnancy and breastfeeding
For reports involving pregnancies where the embryo or foetus could have been exposed to one of your medicines, either through maternal exposure or transmission via semen following paternal exposure, you should:
- make reasonable attempts to follow up all individual cases and collect information on the outcome of the pregnancy and development of the child after birth. For consumer reports, try to follow up with the patient’s healthcare professional.
- collect as much information as possible so you can assess the causal relationship between any reported adverse event(s) and exposure to the medicine.
- consider whether a medicine may have been taken prior to conception or during pregnancy; take into account whether an active substance or one of its metabolites has a long half-life.
You must:
- report pregnancies that result in abnormal outcomes suspected to be related to the medicine as serious adverse reactions. Such cases include:
- congenital anomalies or developmental delay in the foetus or the child
- foetal death and spontaneous abortion
serious adverse reactions in the neonate.
Note: A premature delivery (i.e. earlier than 37 weeks) is not considered an abnormal outcome unless it resulted in adverse reactions to the neonate or mother.
- report suspected serious adverse reactions in infants following exposure to a medicine in breastmilk in accordance with the reporting requirements for serious adverse reactions.
- report any signal of a possible teratogenic effect, such as a cluster of similar abnormal outcomes, as a significant safety issue.
You must submit two separate reports to us if both the parent and the child or foetus experienced adverse reactions.
Consumers or healthcare professionals may contact you for information about the teratogenicity of your medicine and/or experience of its use during pregnancy. You should make reasonable attempts to determine whether any possible exposure of an embryo or foetus to the medicine has occurred and follow up to collect information on the outcome of pregnancy and development of the child after birth.
Unless stipulated as a condition of registration, do not routinely report individual cases of:
- induced termination of pregnancy where there is no information on congenital malformation
- exposure during pregnancy where the outcome is normal or there is no data on outcomes.
You must include these cases in the next PSUR, if one is required, together with aggregated data on overall pregnancy exposure and details of normal and abnormal outcomes. We may request a report from prospective pregnancy registries to be included in a PSUR for evaluation.
Reports of use in paediatric or elderly populations
It is important to collect safety information in paediatric or elderly populations to help identify potential safety signals specific to particular age groups.
For serious adverse reaction reports, you should therefore make reasonable attempts to obtain and provide to us the patient’s date of birth, age or age group.
Lack of efficacy reports
You should record and, if incomplete, follow up all reports of a lack of therapeutic efficacy. You do not need to routinely report individual cases of suspected lack of efficacy. However, you MUST retain the reports made to you and provide them to us if we request them.
There are certain circumstances when reports of unusual or unexpected lack of efficacy must be treated as serious adverse reactions for reporting purposes—for example, where there is a lack of efficacy of:
- medicines used for critical conditions or life-threatening diseases
- vaccines
- contraceptives
- anti-infectives.
You should use your clinical judgement when considering if reports of a lack of efficacy qualify for reporting. You should determine whether the reported lack of efficacy is related to the medicine rather than to an inappropriate treatment or the progression of a disease.
For example, do not report lack of efficacy of antibiotics used in life-threatening situations where the medicine was not appropriate for the infective agent. However, you must report any cases of life-threatening infection where the lack of efficacy seems to be due to the development of a newly resistant strain of bacterium, previously regarded as susceptible, as serious adverse reactions.
Do not report incidences of unexpected lack of efficacy to us if the reporter specifically states the outcome was due to the progression of a disease and not related to the medicine. However, if the reporter believes the outcome was not due to disease progression, this must be reported even if you disagree. You should include your opinions in your report to us.
When you report a suspected lack of efficacy, do not code for the indication for which the suspected medicine was administered as an adverse reaction. For example, do not code hypertension as an adverse reaction to an anti-hypertensive medicine. Rather, where the existing condition was altered—that is, it progressed, recurred or was aggravated—by the lack of efficacy, this should be coded as such in the report.
Reports of lack of efficacy may help identify:
- changes in the manufacturing quality and compliance with good manufacturing practice (see Reports of quality defect issues)
- differences in how a particular subgroup of patients responds to the medicine
- in vaccines, reduced immunogenicity in a sub-group of vaccines, waning immunity and strain replacement
- for anti-infectives, the development of resistance.
If you suspect any of these potential signals, you must report them to us as either an SSI or OSI. Use your professional judgement to determine the seriousness of the issue and whether further investigation and prompt action is warranted.
Reports of use in paediatric or elderly populations
It is important to collect safety information in paediatric or elderly populations to help identify potential safety signals specific to particular age groups.
For serious adverse reaction reports, you should therefore make reasonable attempts to obtain and provide to us the patient’s date of birth, age or age group.
Reports of quality defect issues
You should have a system in place to ensure that you investigate reports of suspected adverse reactions related to quality defects in a medicine promptly (with the urgency indicated by the nature of the defect) and that you notify us of confirmed quality defects or safety issues warranting recall with the least possible delay, in line with the Procedure for recalls, product alerts and product corrections (PRAC) — which covers market action and recall action pathways. You may need to take urgent action in response to a quality defect to protect public health—for example, to recall one or more defective batches of a medicine from the market or issue communications to healthcare professionals.
You must report all serious suspected adverse reactions associated with a suspected or confirmed quality defect in your medicine, including adulterated or counterfeit medicines, in line with the corresponding reporting requirements for serious adverse reactions.
You must also report safety issues related to a suspected or confirmed quality defect to us, for example a possible batch contamination issue or out-of-specification from a stability program. Use your professional judgement to determine the seriousness of the issue and whether it has a potential major impact to the benefit-risk balance of the medicine and public health – if so, it should be reported as a defect AND an SSI.
Reports of transmission of an infectious agent
The transmission of an infectious agent via a medicine is also considered a serious adverse reaction for reporting purposes.
Transmission of an infectious agent may be suspected based on the temporal relationship, clinical signs or symptoms, or on laboratory findings that indicate an infection in a patient exposed to a medicine. Concentrate on the detection of infections or infectious agents known to be potentially transmissible via a medicine, but also consider the occurrence of unknown agents.
When you evaluate any suspected transmission of an infectious agent via a medicine you should, whenever possible, discriminate between the cause (e.g. an injection or other administration) and source (e.g. contamination) of the infection, and the patient’s clinical condition when they were infected (e.g. immunosuppressed, recently vaccinated etc).
Contamination of the medicine concerned, including inadequate inactivation or the attenuation of infectious agents as active substances, is evidence of the transmission of an infectious agent and may suggest a quality defect issue.
Reports of overdose, abuse, off-label use, misuse, medication error or occupational exposure
You MUST report all serious adverse reactions and safety issues in accordance with the prescribed reporting requirements that relate to:
- overdose (accidental and intentional)
- abuse
- off-label use
- misuse
- medication error
- occupational exposure.
You should routinely follow up these cases to ensure you have as much information as possible about:
- early symptoms
- treatment
- outcome
- the context of the adverse reaction (e.g. prescription errors, administration, dispensing, dosage, unauthorised indication or population).
If there is no associated adverse event (i.e. the patient is asymptomatic) or the adverse reaction is not serious, you do not need to report these to us. However, these must be recorded and should be considered in the ongoing review and analysis of the safety of the medicine, as well as in any applicable PSURs. Additionally, these must be provided on request by the TGA within the requested timeframe.
Reports related to orphan drugs
Standard reporting requirements, and any other specific requirements that have been applied as a condition of registration, also apply to orphan drugs.
You must report all serious adverse reactions and safety issues related to orphan drugs in line with reporting requirements.
Reports related to suspended or discontinued products
You must continue to collect and report suspected serious adverse reactions and safety issues for suspended medicines as per the requirements for any approved medicine in Australia.
If a medicine has been removed from the ARTG, you must report serious adverse reactions occurring after a medicine was discontinued until the last batch expiry date. After this date, you should report any new significant follow-up information on reported cases and continue to collect (but not report) safety information for review of delayed onset adverse reactions or retrospectively notified cases.
Any medicines that are supplied after they have been removed from the ARTG (and after the date of final batch expiry) will adhere to the reporting requirements for unapproved medicines as appropriate. See Reporting requirements for medicines supplied through an exemption scheme.
Reports related to Australian product marketed overseas
For medicines on the ARTG not currently marketed in Australia but marketed in other countries, you should ensure there are procedures in place to collect and analyse any information on suspected adverse reactions.
You are not required to report individual adverse reactions that occur overseas to us, but you should include them in your ongoing monitoring activities. You must report safety issues to us. Use your professional judgement to determine the seriousness of each safety issue and whether it qualifies for reporting as an SSI or OSI.
Reports related to overseas products marketed in Australia
Some medicines that are registered or listed on the ARTG and marketed in Australia are manufactured and marketed in different regions by different sponsors overseas. Where there is an agreement in place with an overseas sponsor for such an arrangement, there should be a provision for the timely exchange of safety data in order to monitor the benefit-risk balance of the medicine.
You are not required to report individual adverse events that occur overseas to us, but you should include them in your ongoing monitoring activities. You must report safety issues to us. Use your professional judgement to determine the seriousness of each safety issue and whether it qualifies for reporting as an SSI or OSI.
Your record-keeping requirements
Under paragraph 28(5)(ca) of the Act, you must retain records pertaining to the reporting requirements and safety for your medicine. Information relating to pharmacovigilance activities and the safety of the medicine include, but is not limited to, all adverse reaction reports (serious and non-serious), information surrounding safety issues, special situation reports, reference safety documents and non-valid reports containing drug-event pairs. These must be retained indefinitely for the life of the medicine and for:
- a period of 10 years after removal from the ARTG for registered medicines, and
- a period of 5 years after removal from the ARTG for listed medicines.
General safety information on your medicine, including but not limited to ongoing monitoring activities, PSURs, literature reviews, contracts with pharmacovigilance providers, documentation regarding changes to reference safety information, pharmacovigilance procedural documents and pharmacovigilance training documents should also be retained indefinitely for the life of the medicine. We may also ask to review these records on request, or as part of the Pharmacovigilance Inspection Program.
Your pharmacovigilance system
You must establish and manage a pharmacovigilance system to help you meet your pharmacovigilance responsibilities.
We require you to have an effective pharmacovigilance system in place in order to:
- meet all pharmacovigilance requirements described in these guidelines and applicable legislation
- take any additional pharmacovigilance and risk minimisation actions required by the RMP (if a RMP is in place)
- investigate and report product quality issues associated with serious adverse reactions and safety issues
- critically analyse adverse events and other safety and quality information
- take any action necessary to mitigate an identified safety issue.
Qualified person responsible for pharmacovigilance in Australia
You should have a qualified person responsible for pharmacovigilance undertakings in Australia. Ideally, this person will also be the A-PVCP responsible for reporting to the TGA and coordinating pharmacovigilance-related communications between us. The qualified person responsible for pharmacovigilance in Australia (QPPVA) should ensure that the sponsor has an effective pharmacovigilance system in place and complies with the legal pharmacovigilance requirements.
We recommend the QPPVA:
- lives in Australia
- is permanently and continuously available (or at least within the hours of 9am-5pm AEST Monday to Friday), with a back-up person nominated should the primary QPPVA be absent
- is trained and experienced in pharmacovigilance and relevant legislation in Australia
- is medically qualified, or if not, have ready access to a medically qualified person for any clinical assessments necessary. We prefer that this medically qualified person reside and are medically registered in Australia so they can address adverse reactions, safety issues and the benefit–risk balance of medicines in the Australian context.
Please note that the above are recommendations. Ultimately, the QPPVA should be suitably experienced and qualified in order to monitor the safety of your medicines. The characteristics and skills of the individual QPPVA should be dependent on their specific roles and responsibilities and should ensure that you are able to meet your pharmacovigilance requirements.
The QPPVA needs to have adequate understanding of the Australian and global pharmacovigilance processes to allow them to have effective oversight of the entire pharmacovigilance system.
Pharmacovigilance operational documents and activities
Written procedures
Develop clear written standard operating procedures (SOPs) for pharmacovigilance to ensure all roles, responsibilities, requirements and timelines are well-defined and understood by all personnel involved.
You should make provisions for regular review for improvements to the system where required.
Where you have identified a need to initiate a safety-related update to the reference safety information (that is not a significant safety issue), we expect you to submit safety related updates to reference safety information documents including the Australian PI within a timely manner of identifying the need for change. This ensures safety information available to healthcare professionals and consumers is up-to-date to ensure safe use of your medicines.
Where you have a global parent company, you need to be confident that your SOPs will ensure you become aware of international safety information and regulatory actions in a timely manner. This also applies to activities contracted to third parties. You should review third party procedures to verify that they are adequate and comply with applicable requirements (see Safety contracts and agreements).
Safety contracts and agreements
As a sponsor you are ultimately responsible for complying with Australian regulatory reporting requirements. You are responsible for the ARTG entries you hold and thus, you must ensure you meet your pharmacovigilance responsibilities where you have contractual arrangements with other sponsors or external organisations. This also applies to overseas companies that belong to your parent company, and to other companies within or outside Australia with which you have commercial or contractual agreements—for example, vendors, partners and contract manufacturers.
Where you have contractual arrangements with a third party (external) person, other sponsors or external organisations, you should have a detailed pharmacovigilance contract or agreement in place with them that stipulate the explicit roles and procedures for collecting and reporting safety information to ensure you can comply with your reporting obligations. Further, external companies should be appropriately trained and overseen.
Your pharmacovigilance contract or agreement should specify what safety information is to be collected and how it is to be exchanged, including timelines, reconciliation and reporting responsibilities. Your procedures should prevent you submitting duplicate reports to us.
Training
Staff undertaking pharmacovigilance activities should be appropriately trained in relevant legislation and guidelines, and in the report process and evaluation. Other staff that might receive or process safety reports—for example, clinical development, sales, medical information, legal or quality control staff—should also be trained in collecting and reporting adverse reactions. This ensures that adverse events notified directly to these employees would be reported and communicated through to the pharmacovigilance section for inclusion in the company safety database and for ongoing safety review of a product.
Pharmacovigilance training should be conducted at induction of employment, with an annual refresher at a minimum for all relevant staff as appropriate based on their roles and responsibilities. Training activities undertaken by your staff should be recorded and we may request these documents as evidence of training at any time.
Training recommendations should also apply to external vendors and partners engaged in pharmacovigilance or who have the potential to identify adverse events—for example, market research service providers. Training on the collection and reporting of adverse events should be clearly stipulated in contractual agreements and audited regularly.
Monitoring and collecting information
Your pharmacovigilance system MUST meet any applicable record-keeping and reporting requirements. In terms of monitoring and collecting safety information, your pharmacovigilance system should allow you to:
- identify and collect all information related to the safety of your medicine from all possible sources, including
- spontaneous reports of adverse reactions (including consumer reports to you, or to people who work for you or have a contractual relationship with you)
- internet and social media reports
- reports from non-medical sources
- solicited reports, such as from post-registration studies or post-market initiatives
- reports in international and local literature
- individual adverse drug reaction reports in the TGA’s Database of Adverse Event Notifications (DAEN).
- determine if an enquiry involves an adverse reaction—for example, enquiries to your Medical Information section or sales representatives
- encourage consumers and health professionals to submit safety information—for instance by providing adverse reaction reporting forms and contact details on your website
- gather sufficient information to scientifically evaluate reports of adverse reactions and any other safety issues associated with the medicine
- ensure the reports you collect are authentic, legible, accurate, consistent, verifiable and as complete as possible, and follow these up where necessary
- validate suspected adverse reactions and report them to us within the required timeframe.
Spontaneous reports
All adverse events that healthcare professionals, patients or consumers notify you of, where the reporter suspects a medicine caused the adverse event, are spontaneous reports. Spontaneous reports of adverse events are considered to be adverse reactions for regulatory purposes.
Therefore, you must report these adverse reactions to us if they are considered serious, even if you do not agree with the reporter’s assessment of the cause. A report is only exempt if the reporter specifically states they believe the events to be unrelated or that a causal relationship can be excluded, and you agree with this assessment.
For spontaneous reports where you do not agree there is a reasonable possibility the suspected medicine caused the adverse event, record both your opinion and that of the reporter in your adverse reaction report and provide the criteria you used to make this judgement.
For regulatory purposes, spontaneous reports are considered to have implied causality. That is, where it is not clear whether a causal association is suspected, the medicine and the adverse event are presumed possibly related and therefore meet the definition of an adverse reaction, unless the reporter explicitly states otherwise.
Consumer reports
Reports from consumers are a valuable source of information. You should encourage consumers to report adverse reactions, and to provide mechanisms by which they can do so.
Reports provided by consumers may often lack sufficient clinical detail required for assessing causality or seriousness. Therefore, you should exercise due diligence in ensuring that the reports are complete and are of high quality by accurately recording, clarifying, analysing and following up on any information received.
You should record all reports of adverse reactions from consumers in the same way you would record adverse reaction reports from any other source and consider them when you assess overall safety.
You should obtain as much information as necessary to determine the nature and seriousness of the adverse reaction and seek the reporter’s voluntary informed consent to contact the treating doctor for medical confirmation of the adverse reaction and any additional relevant information.
If you cannot obtain consent, use your clinical judgement to assess how serious the reaction was from the available information and guide your subsequent handling of it. If you deem the reaction to be serious, you should make additional attempts as reasonable either to obtain the reporter’s voluntary consent to contact the treating doctor or ask the consumer to provide relevant medical documentation to allow you to assess causality.
When performing consumer follow-up, if the consumer is not comfortable providing you information, you may give them details for direct reporting of adverse reactions to the TGA.
Further follow-up or medical confirmation may not be necessary for an apparently non-serious adverse reaction.
You cannot disclose the identity of the consumer to us without their explicit consent. You should be familiar with your obligations in relation to the collection, use and disclosure of personal information of consumer reports in accordance with the Australian Privacy Principles and the Privacy Act 1988 and any relevant state or territory privacy legislation, and comply with these obligations.
Internet and digital media reports
The internet, including social and digital media, is an important source of reports of suspected adverse reactions.
You should regularly screen the internet (such as websites, webpages, blogs, vlogs, social networks, internet forums, chat rooms and health portals) or digital media you own, fund, manage or are responsible for, for potential reports of suspected adverse reactions.
You should screen these media often enough to allow you to report valid adverse reactions within the reporting timeframes based on the date of the post.
You should also consider setting up mechanisms for collecting adverse reaction reports through your company website. You can do this by providing reporting forms or contact details for direct communication. You should encourage reports from all sources, including health professionals and consumers.
You do not need to review internet and digital media not sponsored by your company. However, if you become aware of an adverse experience on an internet or digital site that you do not sponsor, you should review the available information and attempt to follow up the report to determine if it must be reported to us.
Reports of adverse reactions obtained from internet or digital media are considered spontaneous and should be evaluated and followed up to determine whether they meet the requirements for reporting.
You must report all valid serious adverse reactions that occur in Australia from company-sponsored internet and digital media to us.
In relation to cases from the internet or digital media, the identifiability of the reporter refers to the existence of a real person.
You should make reasonable attempts to contact the reporter wherever possible to confirm the event and patient details and collect any additional information. For serious adverse reactions, you may post to a public forum/environment and request that the reporter contact you privately to provide more information.
If you do not know what country the primary source is from, use the country where the information was received.
You should keep a record of any adverse reaction reports for ongoing monitoring activities.
Reports from other non-medical sources
You should handle reports of suspected adverse reactions from non-medical sources—for example, the lay press or other media—as spontaneous reports, in accordance with the applicable timeframes. You should make reasonable attempt to follow these reports up to obtain the minimum information you need to make a valid adverse reaction report and determine the seriousness of the adverse reaction.
Solicited reports
Solicited adverse event reports can arise from:
- clinical trials
- non-interventional post-registration studies
- other post-marketing initiatives such as, but not limited to, patient support programs, product familiarisation programs, market research programs and surveys.
For these organised data collection schemes, you should have a system in place to record and document complete and comprehensive case information on solicited adverse events. These adverse events should be systematically assessed to determine whether they are possibly related to the studied (or supplied) medicines.
You should assess causality for all solicited reports—for example, by applying the World Health Organisation Uppsala Monitoring Centre (WHO–UMC) causality assessment system.
Solicited adverse events that the reporting healthcare professional, the investigator, or you as the sponsor judge to have a possible causal relationship to the medicine are considered suspected adverse reactions and therefore subject to reporting requirements.
Adverse reaction reports from non-interventional studies with no systematic adverse reaction collection protocol (i.e. related to medicines other than studied or supplied medicine) are considered spontaneous or unsolicited.
Reports from international and local literature
The international and local scientific and medical literature are a significant source of information for monitoring the safety profile and benefit-risk balance of medicines, particularly in relation to the detection of new safety signals or emerging safety issues.
You should:
- undertake regular—no less than weekly—systematic literature review of widely used reference databases such as Medline, Excerpta Medica or Embase, including those that contain the largest number of articles about the medicine (all active ingredients) and its properties. You should use both trade name and active ingredient names of the medicines in your search strategy.
- monitor ongoing safety and efficacy studies, including non-human teratogenicity and/or carcinogenicity studies for any relevant safety findings
- review and assess both worldwide and relevant local scientific and medical literature articles (including abstracts from meetings and draft manuscripts) to identify, report and record adverse reaction reports and safety issues.
Your search strategies should be documented and reproducible. Your procedures for monitoring literature should sufficiently capture up-to-date and comprehensive safety information associated with your medicines. Specific journals reviewed should be relevant to your product range and should capture any relevant articles not included in the systematic literature review.
Where you make a contractual arrangement with a person or organisation to search the literature, you should ensure that you have a detailed agreement that allows you to comply with your reporting obligations.
For adverse reactions associated with your medicine that are reported in scientific and medical literature, you should:
- identify which adverse reactions occurred in Australia
- identify a single patient
- review and assess the information in the publication.
If the literature references multiple medicines, you should only consider those the author suggests have at least a possible causal relationship with the suspected adverse reaction.
Authors are the primary sources for adverse reactions reported from literature. You should follow up and validate any serious adverse events that are reported in the literature by contacting the study’s author to obtain further information where possible—specifically any information needed to assess causality and patient identifiers.
Do not submit to us any literature reports of adverse reactions that occur in a cohort of patients but do not identify a single patient who experienced an adverse reaction (where reasonable attempts at obtaining this information have not been successful).
Reports from the TGA
Do not routinely report adverse reactions to your medicine that you learn of from us—for example, by interrogating the TGA Database of Adverse Event Notifications (DAEN)—as individual adverse reactions. We encourage you to consult the DAEN regularly to ensure your database is as complete as possible and include these in ongoing signal investigation activities, and reassessment of the benefit-risk profile, including PSUR/PBRERs if required.
However, if the adverse reaction reports could lead to a change in the benefit-risk balance of the medicine, then you must notify us of this change as a significant safety issue.
Analysing information
Pharmacovigilance requires that you not only collect information on adverse reactions and safety issues, but that you also critically analyse and evaluate these to monitor the ongoing benefit-risk profile of your medicine.
Signal detection
Safety issues (or signals) may arise at any stage in the life cycle of a medicine, including the clinical development, manufacturing or in the post-market setting.
You may become aware of safety issues from one or multiple sources which may suggest a new risk or a change in the nature of a known risk associated with your medicine. Safety monitoring activities should include a review of cumulative cases, in order to allow for a comprehensive review of potential safety issues.
You should have a system in place for detecting and investigating such issues in a timely manner.
Signal assessment
You should actively investigate and assess signals you think may indicate a true causal association to determine whether they can be verified or refuted. You should also decide whether any regulatory action is warranted based on your assessment of the risk.
If you verify a signal and after assessment determine additional risk mitigation measures are required, you must report it to us as a safety issue together with any actions you propose to take, or justification for no further action.
Practical Aspects of Signal Detection in Pharmacovigilance: Report of CIOMS Working Group VIII is a useful resource on this.
Data quality control
Data accessibility
The pharmacovigilance data you collect, collate and electronically store must be available from a single access point within Australia.
Data security
You should be familiar with, and discharge, your obligations in relation to the collection, use and disclosure of personal information in line with the Australian Privacy Principles under the Privacy Act 1988 and relevant state or territory privacy legislation.
Electronic data and paper reports of suspected adverse events should be stored and treated in the same way as other medical records, with appropriate confidentiality for information that identifies the patient, reporter or healthcare professional, and in accordance with privacy laws.
You should apply strict access controls to documents and databases to ensure pharmacovigilance data remains secure and confidential across the complete data path.
TGA uses your organisation’s TBS information to determine whether to release information about your products to a person, including pharmacovigilance data. All personnel authorised in TBS can access data held by the TGA about your products. Your nominated TBS administrator must keep your personnel’s contact details up to date, as outlined in the TBS terms and conditions.
You should implement procedures to ensure data is secure and remains uncorrupted when transferred within the company or between any third-party organisations.
Data transfer and validation
You should have mechanisms in place to ensure that, when you transfer pharmacovigilance data within an organisation or between organisations, all data is fully received. Routine reconciliation of data (for example, adverse reaction cases received from post-registration initiatives) should be undertaken, preferably monthly, to ensure the integrity of data transfer and therefore the accuracy of company-held safety information.
All electronic data should have an audit trail. It should be possible to trace all data entry, modification and deletion, including:
- dates and sources of received data and changes
- dates and destinations of transmitted data.
Conformity of stored data with initial and follow-up reports should be verified by quality control procedures, which permit for the validation against the original data or images thereof. Source data—such as letters, emails, records of telephone calls that include details of an event, or an image of the source data—should be easily accessible.
Source paper documents may be transferred into an electronic copy (and vice versa). You should ensure documents are appropriately validated such that all of the information present in the original format is captured and in a legible manner. We do not require that both electronic and hardcopy versions of the original are retained. Source paper documents can be subsequently destroyed provided a confirmed true and complete electronic copy (with back-up) exists.
Data entry
Data should be unbiased and unfiltered. Avoid inferences and imputations when recording the information, and include the primary source’s verbatim text, or an accurate translation, wherever possible.
Adverse events should be coded by a consistent reproducible method, preferably using the appropriate lowest level terms from the Medical Dictionary for Regulatory Activities (MedDRA).
Staff entering data should be trained in the use of the adverse event terminology, and you should confirm their proficiency.
You should verify data is being entered correctly through quality assurance auditing, either systematically or by regular random evaluation.
Data retention
Records of source documents can be stored either in paper or in electronic form. The storage medium selected should be suitable for the required period of retention (i.e. will remain easily accessible and readable over time). Furthermore, consider security and access controls due to the confidential nature of the data and ease of retrieval, as you need to provide requested information to us within the specified timeframes.
Commercially confidential information
Adverse reaction reports and other documents submitted to us may contain information that is commercially confidential.
Please refer to Treatment of information provided to the TGA to see how we manage your information.
Managing duplication
You should have internal processes to identify and manage duplicate cases at data entry and when generating aggregated reports.
Pharmacovigilance and the law
Your pharmacovigilance responsibilities outlined in this guidance are underpinned by legislation.
- Under subsection 28(5)(e) of the Therapeutic Goods Act 1989 and Regulation 15A of the Therapeutic Goods Regulations 1990, you must comply with any reporting requirements that have been prescribed as a condition of registering or listing your therapeutic good in the ARTG.
- Subsection 28(2B) and 28(3) of the Act allows us to impose certain conditions of listing or registration on a therapeutic good when it is entered in the ARTG or at any time while it remains on the ARTG, including when it is suspended (see subsection 29G of the Act). We can use this subsection to require you to submit Periodic Safety Update Reports (PSURs).
- Under Subsection 28(5)(ca) of the Act and Regulation 15A of the Therapeutic Goods Regulations 1990, you must comply with any record-keeping requirements that are prescribed.
- Under Sections 29A and 29AA of the Act, you must advise the TGA in writing as soon as you become aware of information that:
- contradicts information you have already furnished under the Act
- indicates that using the registered or listed medicine in accordance with recommendations for its use may have an unintended harmful effect
- indicates the registered or listed medicine, when used in accordance with the recommendations for its use, may not be as effective as information submitted or relied on at the time of listing or registration would suggest
- indicates the quality, safety or efficacy of the registered or listed medicine is unacceptable.
- Under Subsection 31(1) of the Act, you are required to provide information or documents relating to the safety, efficacy and quality of the goods in the time and format specified by us.
- You must abide by the Australian Privacy Principles set out in the Privacy Act 1988 and any applicable state and territory privacy legislation when you collect, use or disclose personal information.
Our requirements do not override any applicable privacy laws. Any personal information you collect may be disclosed by consent or where the disclosure is required by, or authorised under, a law (e.g. under section 61 of the Act).
Where a report relates to vaccine events, the patient or reporter’s personal information may be disclosed to state and territory health agencies under subsection 61(3) of the Act.
General privacy information is available on our website.
Failure to comply with requirements
- Under subsections 29D(1)(b) and 30(2)(c) of the Act we can cancel or suspend medicines from the ARTG for refusing or failing to comply with a condition of registration or listing.
- Section 21A of the Act specifies the grounds for prosecuting offences related to not complying with conditions of registration or listing.
- Section 29A of the Act specifies the grounds for prosecuting offences related to failing to notify us of adverse reactions and safety issues related to your medicine.
Other pharmacovigilance requirements
European Union (EU) pharmacovigilance guidelines currently adopted in Australia can be found on the TGA website. We commonly reference guidance documents published by the EU where required and appropriate.
Pharmacovigilance Inspection Program
In line with other international regulators, TGA conducts inspections to assess whether sponsors are meeting their Australian pharmacovigilance regulatory obligations, including those specified in these pharmacovigilance guidelines.
The Pharmacovigilance Inspection Program (PVIP) applies to all sponsors of listed and registered medicines. For more information, please see the PVIP Guidelines.
Periodic Safety Update Reports
A Periodic Safety Update Report (PSUR) is a systematic review of the global safety data of an approved medicine that becomes available to you during a defined time period. PSURs are also referred to as Periodic Benefit-Risk Evaluation Reports (PBRERs).
PSURs are required for certain registered medicines. The requirement to submit a PSUR can be applied as a condition of registration under section 28(2B) of the Act when the medicine is included on the ARTG. PSURs are not required for all medicines, only those on which this specific condition is imposed. This means that we are not able to provide general advice on relevant PSUR requirements because these may vary depending on the specific conditions of registration for a particular medicine.
There is an internationally agreed format for producing a PSUR. A PSUR should present a comprehensive and critical analysis of the benefit-risk balance of a medicine and take into account new and emerging significant safety findings, in the context of cumulative information on benefits and risks.
For more information on PSURs, refer to the EMA guideline EMA/816292/2011 Rev 1* (9 December 2013) Guideline on good pharmacovigilance practices (GVP) Module VII – Periodic safety update report with TGA annotations and the ICH guideline E2C (R2) on periodic benefit-risk evaluation report (PBRER) (CHMP/ICH/544553/1998).
Risk Management Plans
A Risk Management Plan (RMP) documents the risk management system required to identify, characterise and minimise a product’s important risks. This typically comprises both the EU RMP and an Australia-specific annex (ASA).
RMPs are required for certain higher-risk applications to enter a medicine in the ARTG or to vary an ARTG entry. Throughout the lifecycle of the product, RMPs must be maintained, and important updates submitted to the TGA for evaluation. The requirement to implement a RMP and subsequent versions agreed by the TGA can be applied as a condition of registration under section 28(2B) of the Act when the medicine is included on the ARTG.
For more information on RMPs, refer to Risk management plans for medicines and biologicals Australian requirements and recommendations.
Black Triangle Scheme
Inclusion in the Black Triangle Scheme is imposed as a condition of registration under section 28(2B) of the Act for certain prescription medicines included on the ARTG. Sponsors of medicines included in the Black Triangle Scheme are required to comply with the actions outlined in the Black Triangle Scheme: Information for sponsors for the required duration of inclusion in the scheme.
Reporting requirements for medicines supplied through an exemption scheme
When a medicine is used because access has been granted through an exemption scheme, such as the Special Access Scheme, Authorised Prescriber Scheme or Clinical Trials Scheme, different reporting requirements apply.
Special Access Scheme
The procedures for reporting adverse reactions to medicines accessed under the Special Access Scheme are outlined in Special Access Scheme: Guidance for Sponsors.
Authorised Prescriber Scheme
The procedures for reporting adverse reactions to medicines accessed under the Authorised Prescriber Scheme are outlined in Authorised Prescriber Scheme: Guidance for medical practitioners, Human Research Ethics Committees, specialist colleges and sponsors.
Clinical Trials Scheme
For adverse reaction reports and safety issues that arise from Australian clinical trials where the medicine is used outside the approved indications or intended use, refer to Clinical trials and the Note for guidance on clinical safety data management: definitions and standards for expedited reporting.
Reporting requirements between application submission and prior to inclusion in the ARTG
In the period between the submission of a registration application and the decision to register the medicine, you are not required to routinely submit individual adverse reaction reports except when the medicine is being used in Australia in a clinical trial (in accordance with the Clinical trial guidelines).
However, during this pre-registration period you may receive information that impacts on the benefit-risk balance from countries where the medicine is already marketed, from use in ongoing clinical trials, or from use on a compassionate basis. These may be identified during ongoing review and analysis of all information. This information includes foreign reports of adverse reactions, information about the safety or benefit-risk profile of the medicine, or action taken by a foreign regulatory agency, and the basis for such action.
You are encouraged to report this as a safety issue to the relevant pre-marketing area of the TGA.
You should use your clinical judgement to determine what constitutes a change to the benefit-risk balance. For example, normally another report of a well-known adverse reaction would not be considered significant, but a report of an unexpected or new serious suspected reaction with good evidence of a causal relationship, or where there is suspicion of a change in the frequency or severity of a known effect, would be considered relevant to the evaluation. Similarly, results from studies that impact on the assessment of efficacy would be considered significant. You may be required to justify a decision not to report.
For any safety issues for a prescription medicine with an ongoing application but not yet registered on the ARTG, notify the relevant TGA Case Manager via streamlinedsubmission@health.gov.au.
For any safety issues for a non-prescription medicine with an ongoing application but not yet registered or listed on the ARTG, notify otc.medicines@health.gov.au or complementary.medicines@health.gov.au, as appropriate.
Lapsed or withdrawn applications
When an application to include a medicine on the ARTG lapses or is withdrawn, the Secretary of the Department of Health and Aged Care may require you, under section 29B of the Act, to disclose whether you are aware of certain information mentioned in subsection 29A(2) or 29AA(2) about the medicine and, if so, to provide that information.
Appendix – Safety issue reporting decision tree
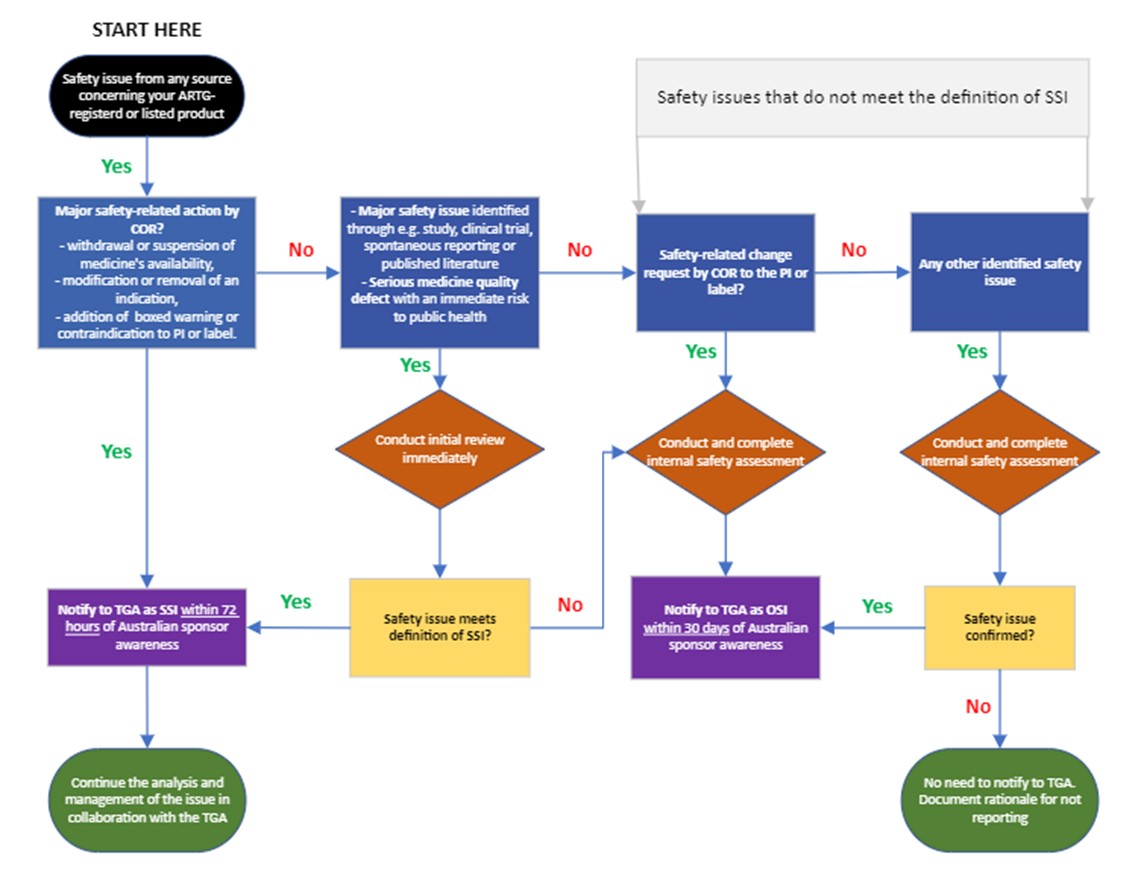
{kind=link}
This flowchart outlines the process for handling safety issues related to ARTG-registered or listed products. It starts with identifying a safety issue from any source.
The chart then branches into different paths based on the nature and severity of the issue:
- If there's a major safety-related action by COR (like withdrawal of medicine, modification of indication, or boxed warning or contraindication), notify TGA as SSI within 72 hours.
- If no major COR action, but a major safety issue is identified through studies, trials, or literature, conduct an immediate initial review. If it meets the definition of SSI, notify TGA within 72 hours. If not, proceed to the next step.
- If there's a safety-related change request by COR to the PI or label, conduct an internal safety assessment. Notify TGA as OSI within 30 days if confirmed.
- For any other identified safety issue, conduct an internal safety assessment. If confirmed as a safety issue, notify TGA as OSI within 30 days. If not confirmed, document the rationale for not reporting.
The chart emphasises timely reporting to TGA and appropriate internal assessments based on the type and severity of safety issues.
Page history
V3.0 (no version change as only formatting and accessibility changes made). Migration to new 'Guidance' content type included changes to the following:
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
V3.0 Updates included:
- update to safety issue reporting requirements
- major formatting changes
- inclusion of safety issue reporting decision tree and online safety issue reporting form.
V2.2 Minor content update due to inclusion of link to Frequently Asked Questions document.
V2.1 Minor content update due to revised links to online adverse event reporting forms.
V2.0 Updates included:
- new title
- revision to web-based format
- new record-keeping requirements
- new and updated information.
V1.3 Minor updates to pharmacovigilance contact person text in Section 2.1.
V1.2 Minor updates to the healthcare professional definition to remove coroner and overdose definition to remove final qualifying sentence.
V1.2 Update to include details of nominating/updating contact person for pharmacovigilance through eBS Help Desk.
V1.0 Original publication.
V3.0 (no version change as only formatting and accessibility changes made). Migration to new 'Guidance' content type included changes to the following:
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
V3.0 Updates included:
- update to safety issue reporting requirements
- major formatting changes
- inclusion of safety issue reporting decision tree and online safety issue reporting form.
V2.2 Minor content update due to inclusion of link to Frequently Asked Questions document.
V2.1 Minor content update due to revised links to online adverse event reporting forms.
V2.0 Updates included:
- new title
- revision to web-based format
- new record-keeping requirements
- new and updated information.
V1.3 Minor updates to pharmacovigilance contact person text in Section 2.1.
V1.2 Minor updates to the healthcare professional definition to remove coroner and overdose definition to remove final qualifying sentence.
V1.2 Update to include details of nominating/updating contact person for pharmacovigilance through eBS Help Desk.
V1.0 Original publication.