Completing the bioequivalence study information form
Introduction
This document will assist applicants completing the Bioequivalence Study Information Form (BSIF) for inclusion in an application for a new prescription generic medicine. It includes information and instructions on what is required in certain sections of the template.
Bioequivalence related information can be included in different places in a dossier even when following Common Technical Document (CTD) protocol. The BSIF consolidates key bioequivalence information and the locations of critical data in one place.
We will use the completed template to record our evaluation comments. The amended template will then be included as part of the assessment reports.
Completion of the BSIF may also remove the need for you to provide some information in the dossier itself .
Including the BSIF in an application
Inclusion in Module 1 of the CTD
Submit the completed BSIF in your dossier. It must be included in:
- pdf format
- CTD module 1.9.1
The completed BSIF in Microsoft Word or rich text (RTF) formats should be available upon request.
For more information about CTD and dossier requirements, refer to the following TGA webpages:
If you have trouble accessing documents on the TGA website, refer to TGA webpage on accessing documents .
Replacing existing mandatory requirements
Applications for new generic medicines that include a bioequivalence study usually require the following documentation:
- Summary of a Bioavailability or Bioequivalence Study form
- CTD module 2.5
- CTD module 2.7
When you submit a completed BSIF, these documents are generally not needed.
Exception
If the proposed Product Information (PI) contains clinical information that is different to the reference product’s PI, you must provide module 2.5 in addition to the completed BSIF.
Multiple bioequivalence studies
When you need to submit more than one (1) bioequivalence study:
- replicate sections 2 to 9 for each subsequent study
- include the replicated sections at the end of section 9
- Do not complete separate BSIF templates for each study
- Do not update the section numbering for the duplicated sections.
Related templates
TGA biowaiver templates
There are TGA templates for proposed biowaivers that provide the same benefits as the BSIF and can be submitted in conjunction with a BSIF.
- additional strengths biowaiver template
- Biopharmaceutics Classification System (BCS)-based biowaiver template.
For further information on these biowaiver templates, see guidance for Completing biowaiver templates.
Equivalent overseas templates
If you have had an application accepted in Canada, Singapore or Switzerland, and the dossier included any of the following templates:
- Swissmedic Bioequivalence Trial Information Form
- Health Canada draft comprehensive summary - bioequivalence (CS-BE) template
- Australia-Canada-Singapore-Switzerland Consortium Bioequivalence Study Information Form
Then you can include that completed template in your application to the TGA.
It is considered equivalent to a completed BSIF. Include it in your Australian dossier in module 1.9.1 instead of the BSIF.
The reduced CTD requirements will also apply, as detailed in Replacing existing mandatory requirements above.
Completing the BSIF
When you complete the BSIF:
- provide succinct and accurate responses
- check you have included all information
- check you have included correct references.
Do not change the format or content of the template (headings, instructions, requests and tables) unless instructed to do so (see below).
Locating information from the dossier
Many questions in the template request the location of a piece of information within the dossier. The responses to this type of questions must include:
- the module
- the volume
- the tab index
- the page number.
For example:
- BSIF section 2.2 question: 'Location of the principal investigator(s) signed c.v.''
Answer: 5.3.1.2, Appendix 16.1.5, Signature of principal or coordinating investigator, p 2 - BSIF section 2.4.3.1 question: 'Location of the certificate of analysis (CoA)'
Answer: 3.2.P.5.4, Batch analysis, p 23
The use of hyperlinks to the data within the dossier is preferable as this will greatly assist in the evaluation process.
Sourcing evidence from literature
When your response relies on publically available information, published literature or any other third party source, this information must be easily accessible to the evaluators. You must:
- provide a full copy of the information, and
- in your response, state where the full copy is located (refer to instructions above on Locating information from the dossier).
Attaching information
You can add attachments at the end of the template if your response to a question in the BSIF requires further information. Include a link in your response to where the attachment is located in the BSIF by creating a hyperlink.
Caution on the size of electronic documents when submitted via email
Ensure the email containing the completed template is in total less than 30MB For more information, see TGA's general dossier requirements submitted via email
Information needed in responses
Guidance on the type of information you should provide and how you should present it, for some sections in the BSIF, is given below under the corresponding section titles.
Section 1 - Summary
1.1 - Pharmacokinetic properties
Provide pharmacokinetic properties of the drug substance based on publically available information (for example, journals, Public Assessment Reports and Product Information).
Under 'what were the other relevant pharmacokinetic characteristics of the drug substance(s)?' include the following characteristic information relevant to the drug substance:
- permeability (or absolute bioavailability)
- distribution
- metabolism
- mode, route and rate of elimination
- first-pass effect (if any) and its significance
- in vitro interconversion of enantiomers (if any)
- effect of age/food/ genetic polymorphism (if any)
1.2 - Summary of bioequivalence studies performed
The brief description of each comparative bioavailability study must include:
More information regarding the choice of the reference product is requested in Section 2.4.3.3 of the BSIF.
See instructions below for what to provide in Section 2.4.3.3 of the BSIF.
- study number
- study conditions
- identity of the test and reference product used in the study
- bioequivalence acceptance criteria and a scientific rationale for the choice of these criteria
- confirm if the reference product is an Australian reference product or an overseas reference product.
- identity of the substance being investigated (i.e. a parent compound, inactive pro-drug or metabolite). If it is a metabolite, indicate if it is found endogenously or has enantiomers
- if not all strengths were investigated, provide reason for the choice of the strength(s) investigated.
Example response
Study number 1234, open label, randomised, two-period, two-sequence single dose crossover study of 50 mg XXXX tablet (Generic company. Ltd) and 50 mg reference tablet (Reference company. Ltd) under fasting condition. The reference product is the Australian innovator product. The acceptance interval for AUC0-t and Cmax is 90.00-111.11% of metabolite YYYY because the medicine is a narrow therapeutic index product. The measured substance is the main active metabolite YYYY. The active pharmaceutical ingredient (API) is an inactive-prodrug that has immeasurably low plasma concentration that quickly eliminates. The plasma metabolite YYYY exposure reflects parent compound and metabolite formation is not saturated at therapeutic doses (literature citation or data in).
Only the highest strength has been tested as all products in the range have the same proportional API to excipient ratio, and absorption increases linearly within dosage range (10-50mg) under fasting or fed conditions (literature citation).
1.3 - Biowaivers for strength(s) not tested in bioequivalence studies
Under 'are these product strengths systemically active immediate release oral dosage forms?', if you answer 'No', you need to make sure that the documents mentioned in the space provided contain information that justifies waiving the bioequivalence studies for these additional strength products, i.e. biowaiver justification.
Our regulatory approach to therapeutic products closely align with those of comparable international regulatory counterparts wherever possible. Therefore, consider the following international guidelines to assist you preparing biowaiver justification for these products other than immediate release oral dosage forms (for example, patches or modified release oral dosage forms):
- Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev 1/Corr**), adopted with annotation
- Guideline on quality of oral modified release products (EMA/CHMP/QWP/428693/2013)
- Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CHMP/EWP/280/96), adopted with annotation
- Questions and answers: positions on specific questions addressed to the Pharmacokinetics Working Party (EMA/618604/2008)
If your approach deviates from a TGA adopted international guideline, ensure to include justification for any deviations as part of the biowaiver justification.
The information you provide on biowaiver justification could be large. Therefore, the evaluation of this information will be provided in another assessment report.
Do not provide information on the use of an overseas reference product in this section. Provide this information in Section 2.4.3.3 of the BSIF. See instructions below on what to provide in Section 2.4.3.3 of the BSIF.
Section 2 - Clinical study report
2.4.1 - Overall study design and plan - Description
The brief description of the overall study design and plan must at least include:
- study sequence
- number of subjects per sequence
- dosing regime
Example responses
- Partial replicate study with sequences TRR and RTR in 48 healthy subjects. The washout is 2 weeks. The dose was 4 actuations of the inhaler equivalent to 100 micrograms. There were two cohorts of 24 subjects with the second only to be used if bioequivalence was not observed after the first.
- Parallel group in 36 patients with a 1 week lead for each period. The dose was one 75 mg tablets each morning and evening as per the dosing instructions in the draft Product Information.
2.4.2.3 - Health verification
Under 'Criteria used and all tests performed to judge study subject health status' list all physical examinations, pathology tests, drug screening and medical history collected pre-study, during study and post-study.
Under 'Health verification schedule or dates when the tests were performed' include location in the dossier of the full schedule in the bioequivalence study report, as relevant.
2.4.2.4 - Subjects enrolled and the removal of subjects
For 'Number of subjects enrolled in the study' provide the following information, as relevant:
- number of subjects estimated
- how the subject sample size was estimated
- number of participating subjects
- total number of subjects who completed the study.
Under 'Do any of the following apply?' if you answer 'Yes' to alternates, withdrawals or dropouts, your reasons should include:
- the subject number (assigned subject identification number)
- at what point the alternate/withdrawal/dropout occurred in the study
- for alternates:
- how alternates were included or excluded from the study
- for withdrawals/dropouts:
- which period of study the withdrawal/ dropout subject had completed, if applicable.
Definition
- Alternates are 'spare' study subjects who are included in the analysis only if needed as replacements for excluded subjects.
- Withdrawals are subjects excluded from the study at the discretion of the Investigator
- Dropouts are subjects who excluded themselves from the study by their own discretion, e.g. withdrew consent or did not show up to the study.
2.4.3.2 - Reference product
For 'Potency (assay, % label claim)' the reported assay result must be obtained using the same conditions as the test product
2.4.3.3 - Justification of the choice of reference product
Under 'Justify the choice of reference product' confirm that the reference product has been chosen because it is the Australian reference product. Provide location of the email correspondence with TGA on the choice of Australian reference product, if applicable.
If the reference product is not the Australian reference product (overseas reference product used), then:
- state the country in which it is marketed
- include a justification for its suitability to be used as the reference product
- include location in the submission of the full details, including analytical tests, demonstrating suitability.
For further information refer to TGA guidance on Biopharmaceutic studies - choice of the reference product for bioequivalence of generic medicines.
Under 'Location of the reference product confirmation evidence' for 'Photographic images of the reference product carton and primary container labels' the images provided must clearly show:
- name of the product
- marketing authorisation holder
- name and address of the manufacturer
- batch number
- expiry date.
2.4.4 - Selection of doses in the study
For 'How many dosage units comprise a single administered dose?' if your answer is for systemically active non-unit dosage forms where the delivered dose is estimated based on subject and treatment (such as oral suspensions, inhalations, sprays, creams), provide the estimated delivered dose.
Example response
2.5 mg/kg bodyweight/day
2.4.6.2 - Sampling protocol
For Deviations from the sampling protocol' the response should address the following:
- description and explanation for deviations
- impact of these deviations on the study
- whether deviations from the nominal sampling times were accounted for in the pharmacokinetic analysis.
Section 3 – Study subjects
3.1 - Demographic and other baseline characteristics
For 'Study population' state if study subjects are, for example, normal healthy adult volunteers or paediatric patients.
For 'Subjects with special characteristics' identify the subject number and state notable characteristics.
Example response
Subject 05 was a fast acetylator of debrisoquine
Section 4 – Protocol deviations
In this section, you do not need to repeat information regarding deviations to the sampling protocol. This is covered under section 2.4.6.2 of the template.
Section 5 – Safety evaluation
Under 'Were there any adverse events following administration of the test or reference product?', if you answer 'Yes', your response addressing 'observed adverse events' includes:
- subject number who showed the adverse reaction
- the adverse event
- when did the adverse event occurred (for example, following administration of the test product)
- any causal relationships
- required treatments and outcomes
- the implications of the observed adverse events with respect to bioequivalence.
Section 6 - Efficacy evaluation
6.2.1 - Calculation of pharmacokinetic parameters
Under 'How were the pharmacokinetic parameters calculated/ obtained for AUC0-inf, AUC0-t, C max, t max, the elimination rate constant, and t½?' provide the following:
- detail on how it was calculated, i.e. using the linear, log or log/linear trapezoidal rule
- for AUC AUC0-inf, how many points were used to calculate λ.
6.2.2 - Pharmacokinetic (PK) parameters results
Choose one of the following options to provide PK parameter results:
- Complete the relevant table for single dose or multiple doses. You may modify as necessary by:
- adding extra tables for different APIs or where there is data a metabolite.
- deleting the unused table.
Provide the unit of measurement in the 'Parameter' column after each PK parameter. For example, AUC0-t (ng.hr/mL).
- You may replace these tables with ones you have included in module 5.3.1 if they contain the same requested information. The replaced table should not be an image or a picture.
6.3.1 - Statistical analysis calculation
Under 'Was the statistical analysis method different to as described in the TGA adopted EU guideline?'', if you answer 'Yes' provide the following:
- detail on the fixed and random effects.
For example, for a two stage study these should be stage, sequence, sequencestage, subject (sequencestage), period (stage) and formulation - the applied confidence interval.
For example, using 94.12% confidence intervals for both the analysis of stage 1 and the combined data from stage 1 and stage 2 for a two stage design.
6.3.2 - Geometric means, results from ANOVA, Degrees of Freedom (DF) and intra-subject derived coefficient of variation (CV)
Follow the instructions given previously for section 6.2.2 PK parameter results. The results provided for Test and Reference must be the geometric means.
6.3.3 - Comparison of the results
Under 'How did the study results compare with the publicly available data of the reference product and pharmaceutically equivalent products (if any), including mean values, inter- and intra-individual variability?' addresses the following your answer:
- compare the mean results of the current study to those of the reference product and pharmaceutically equivalent products (if any) in the literature for C max, T max, AUC0-t, AUC0-inf and t1/2
- comment on the inter- and intra- subject variabilities
- include links to the papers relating to these BE studies. If none found, state none found. The journals, Public Assessment Reports and Product Information should be included in the dossier.
Definition
Pharmaceutically equivalent products are products which contain the same amount of the same active substance(s) in the same dosage forms that meet the same or comparable standards
6.3.4 - Statistical effects
In this section, discuss whether there was any period, subject and /or sequence effects and why these should not compromise the validity of the study.
Section 7 – Analytical validation report
7.1 - Analytical technique
For 'Location of the internal standard CoA' the CoA must include information on isotopic purity, for labelled materials.
7.2 - Selectivity
Provide the methods to verify selectivity against endogenous/exogenous compounds, metabolites and enantiomers (where relevant) and the results.
7.5 - Standard curves
For 'Back-calculated concentrations of the calibration standards of the validation runs' highlight in your response any values outside the acceptance range. For example, ±15%, except ±20% for LLOQ
7.10 - Stability
Your answers in 'summary of the data' column for each stability study should provide the following:
- a description of the methodology employed
- number of individual samples at each concentration exposed to the stability test conditions
- summary of the results
In the last row headed 'Others', identify if other stability studies were performed, such as:
- long-term stock solution and working solution stability
- short-term stock solution and working solution stability
- dry-extract stability
- wet-extract stability
- stability in blood before sample processing
You may add more rows as required.
Section 8 - Bioanalytical study report
8.1 - Analytical technique
For 'Longest period of subject sample storage' identify the time elapsed between the first day of sample collection and the last day of subject sample analysis.
8.2 - Standard curves
For 'Number of curves run during the study for subject sample analyses' include both valid and failed runs, and include the reasons for the failures.
For 'Back-calculated concentrations of the calibration standards of the study runs' highlight in your response any values outside the acceptance range. For example, ±15%, except ±20% for LLOQ.
8.3 - Quality control samples
For 'Back-calculated concentrations of the QC samples of the study runs' highlight in your response any values outside the acceptance range. For example, ±15%.
8.5 - Repeat analysis (re-analysis, re-injection and re-integration)
Under ‘do any of the following repeat analyses apply?’ if your answer is ‘Yes’ to any repeat analyses (i.e. re-analysed samples, re-injected samples or re-integrated chromatogram samples), complete the ‘summary details of repeated samples’ table. You may add more rows as needed.
Alternatively, you may replace this table with the one you have included in module 5.3.1 if it contains the same requested information. The replaced table should not be an image or a picture.
In the first column headed 'Sample number', include any of the following, as relevant:
- subject number
- sample number
- sample collection time/date.
8.6 - Incurred sample reanalysis (ISR)
Definition
The percent difference (% difference) is the difference between the initial concentration and the concentration measured during the repeat analysis calculated in relation to the mean value, see equation below:
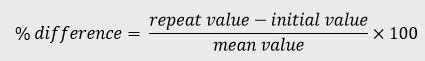
For more information on incurred sample reanalysis, refer to CPMP/EWP/QWP/1401/98 Rev 1/Corr**.
Section 9 - Quality assurance
In this section, tables are included to address internal quality assurance methods (section 9.1) and monitoring, auditing, inspections of the clinical and bioanalytical sites (section 9.2).
- Complete the tables, adding more rows as needed, or
- You may replace these tables with ones you have included in module 5.3.1 if they contain the same requested information. The replaced table should not be an image or a picture.
Reminder
The clinical and bioanalytical sites are those you included in section 2.2 of the template.
Section 11 - Applicant's response to the list of TGA questions
Only complete this section if we have raised questions during the evaluation.
Our questions will be located in section 10 of the template under the space provided for 'TGA use only - List of questions'. You may also find a copy of the questions raised in the consolidated s31 request letter.
If you intend to attach any information as part of your answer to this section, refer to Attaching information above.
Version history
| Version | Description of change | Author | Effective date |
|---|---|---|---|
| V1.0 | Original publication | Scientific Evaluation Branch | December 2019 |
| V1.1 | removed 2.2 | Scientific Evaluation Branch | April 2020 |