Providing biopharmaceutic data for medicine applications
Guidance on bioavailability and/or bioequivalence aspects of medicines including information on biowaivers.
Purpose
This guidance is intended to assist sponsors with applications for market authorisation of medicines that require demonstration of bioavailability and bioequivalence.
This guidance covers:
- all matters relating to bioavailability and/or bioequivalence aspects of medicines containing active substances that are synthetic chemical entities
- active substances that are:
- antibiotics
- short-chain synthetic polypeptides
- some hormones (steroid hormones and synthetic peptides of usually less than 32 amino acids-some exceptions may apply).
This guidance does not cover:
- Matters relating to other pharmacokinetic studies.
Legislation
Adopted European guidelines for biopharmaceutic studies
The guidance is to be applied in addition to the following European Union guidelines that have been adopted by the TGA, some with annotations:
- Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev 1/Corr**), adopted with annotation.
- Guideline on quality of oral modified release products (EMA/CHMP/QWP/428693/2013)
- Guideline on the quality of transdermal patches (EMA/CHMP/QWP/608924/2014)
- Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CHMP/EWP/280/96), adopted with annotation
- Guideline on the clinical investigation of the pharmacokinetics of therapeutic proteins (CHMP/EWP/89249/2004).
- Questions and answers: positions on specific questions addressed to the Pharmacokinetics Working Party (EMA/618604/2008).
Comparative dissolution profiles for biopharmaceutic studies
When dissolution profiles or a similar term is used in this guidance, data should be generated in a comparative manner as follows:
- At least 12 dosage units (e.g. tablets, capsules) of each batch must be tested individually, and mean and individual results reported.
- The percentage of nominal content released are measured at a minimum of three (3) suitably spaced time points (excluding zero time point) to provide a profile for each batch (e.g. at 5, 15, 30 and 45 minutes, or as appropriate to achieve virtually complete dissolution).
- The batches are tested using the same apparatus and, if possible, on the same day.
- The stirrer used is normally a paddle at 50 rpm for tablets and a basket at 100 rpm for capsules. However, other systems or speeds may be used if adequately justified and validated.
- Test conditions are those used in routine quality control or, if dissolution is not part of routine quality control, any reasonable, validated method.
- The similarity factor, f2, is calculated using the equation and conditions stated in Appendix I of the European Medicines Agency (EMA) Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev 1/Corr**) to demonstrate the similarity of two dissolution profiles. The f2 value must be between 50 and 100.
- If more than 85 per cent of the active substance is dissolved within 15 minutes in all tested batches, dissolution profiles are considered to be similar without the need to calculate the similarity factor.
- If there are insufficient quantities of recently manufactured batches available to meet this requirement, then both:
- test retention batches
- explain in the test report why this was done, stating the age and storage history of the samples.
Medicines that do not require biopharmaceutic data
We do not require biopharmaceutic data or a justification for not providing this data for:
- medicinal gases
- peritoneal dialysis solutions
- simple aqueous solutions for intravenous injection or infusion (simple solutions do not include complex solutions such as emulsions, micellar or liposomal solutions)
- other parenteral routes, e.g. intramuscular or subcutaneous, provided that the test product is of the same type of solution (aqueous or oily) and contains the same concentration of the same active substance and the same excipients in similar amounts as the reference product
- simple solutions that do not contain a pharmacologically active drug substance e.g., sodium chloride injection
- oral solutions that both:
- contain the same drug substance(s) in the same concentration as a currently registered oral solution
- do not contain excipients that may significantly affect: gastric passage or absorption of the drug substance(s) in vivo solubility or in vivo stability of the drug substance
- medicines containing drug substances that are not systemically or locally absorbed. Examples include:
- barium sulfate enemas
- oral suspensions
- nonbiodegradable ion exchange resins
- other nonbiodegradable long-chain polymers
- powders from which no ingredient is absorbed.
Note
A study or justification may be required if there is doubt as to whether absorption occurs.
Medicines applied locally (e.g. inhalation and nasal medicines, ocular, dermal, rectal, or vaginal administration) except where the drug substance is acting systemically.
Note
Inhaled steroid products should be supported by data on systemic exposure as part of the evidence of their safety, even in cases where they are intended to act locally.
Biopharmaceutic data may be relevant in some circumstances as described in the Guideline on the requirements for clinical documentation for orally inhaled products (CPMP/EWP/4151/00 Rev. 1).
- Medicines with an acceptable correlation between the rate and extent of in vivo absorption and the in vitro dissolution rate, and where the in vitro dissolution rate of the new medicine is equivalent (under the same test conditions used to establish the correlation) to a registered medicine.
- Minor formulation changes to colouring agent, inked imprint, flavour or fragrance, provided the changes are Self-Assessable Requests as outlined in Variations to prescription medicines - excluding variations requiring evaluation of clinical or bioequivalence data: Process guidance.
We require a justification for not providing biopharmaceutic data for all formulation changes to medicines other than those listed in this section (15.3).
Related information and guidance
Medicines that require biopharmaceutic data
We require biopharmaceutic data (unless otherwise justified) for new medicines that are:
- oral tablets
- oral capsules
- oral suspensions
- complex intravenous solutions for injection
- applied locally (e.g., inhalation and nasal medicines, ocular, dermal, rectal or vaginal administration) where the drug substance is acting systemically
- transdermal medicines.
For a new chemical entity
Absolute bioavailability study (compared with an intravenous injection or infusion). In the absence of an absolute bioavailability study, a relative bioavailability (compared with an oral solution or suspension of defined particle size).
Bioavailability studies to determine the relative bioavailabilities of the individual enantiomers in racemic drug substances.
Bioequivalence of the to-be- marketed formulation(s) compared with the formulation(s) used in pivotal dose-defining and efficacy studies.
Bioequivalence among the different strengths of the medicine proposed for registration.
Bioavailability studies to show the effect of food.
For a new salt
Biopharmaceutic data for the active moiety in the new salt compared with the currently registered salt.
For a new fixed combination medicine
Bioequivalence of the drug substance in the fixed combination medicine to each of the registered medicines containing the single entity.
Additional studies as required by the Guideline on clinical development of fixed combination medicinal products (EMA/CHMP/158268/2017).
For a new dosage form
Bioequivalence of the new dosage form to the currently registered dosage form(s).
For a new strength
Bioequivalence of the new strength(s) to the currently registered strength(s) of the innovator medicine if the biowaiver criteria are not met according to either:
- For immediate release dosage forms - Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev. 1/ Corr **)
- For modified release dosage forms - Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CHMP/EWP/280/96).
For a new generic medicine
Bioequivalence of the new generic medicine to the corresponding innovator medicine marketed in Australia.
If the innovator medicine is no longer marketed in Australia, please email streamlinedsubmission@tga.gov.au for advice regarding the appropriate reference product against which the bioequivalence study should be conducted.
For a new formulation that may affect bioavailability
For innovator medicines, bioequivalence of the new formulation to the original formulation, unless justified in line with Variations to prescription medicines - excluding variations requiring evaluation of clinical or bioequivalence data, Appendix 1: Variation types - chemical entities.
For other medicines, bioequivalence of the new formulation to the corresponding innovator medicine, unless justified in line with Variations to prescription medicines - excluding variations requiring evaluation of clinical or bioequivalence data, Appendix 1: Variation types - chemical entities.
For a new modified-release formulation
The appropriate study(ies) in line with the following TGA adopted EMA guidelines:
- Guideline on quality of oral modified release products (EMA/CHMP/QWP/428693/2013).
- Guideline on the quality of transdermal patches (EMA/CHMP/QWP/608924/2014).
- Guideline on the pharmacokinetic and clinical evaluation of modified release dosage forms (EMA/CHMP/EWP/280/96) as amended.
Validation and quality control of assay procedures used in biopharmaceutic studies
Fully describe analytical procedures and conditions of sampling, in the form of a standard operating procedure.
The analytical methods should be specific and adequately sensitive, and preference should be given to chromatographic techniques such as high-performance liquid chromatography (HPLC) or gas chromatography (GC).
Clearly state the criteria for accepting or rejecting assay data in the protocol or study report.
Provide a few examples of chromatograms to demonstrate sensitivity and selectivity of the analytical test methods.
Retain all of the original printouts in case we require them, but do not include every printout in the application.
Related information and guidance
Guideline on the investigation of bioequivalence (CPMP/EWP/QWP/1401/98 Rev 1/ Corr**) briefly outlines the general requirements for validation and quality control of assay procedures used in biopharmaceutic studies, and refers to other EU guidelines on validation of assay procedures.
Guideline on bioanalytical method validation (EMEA/CHMP/EWP/192217/2009)
United States Food and Drug Administration's (FDA's) Bioanalytical Method Validation Guidance for Industry.
Choice of the reference product for bioequivalence of generic medicines
Reference products
To register a new generic medicine in Australia, you must demonstrate bioequivalence against the Australian reference product.
A bioequivalence study using an overseas reference product may be acceptable, provided you can demonstrate identicality between the Australian and overseas reference products.
An overseas reference product can be used for:
- solid oral, immediate and modified release tablets and capsules
- oral suspensions.
For inhalation products see guidance on inhalation and nasal spray registered medicines.
The Australian reference product should be used for other dosage forms that require a bioequivalence study (see Medicines that require biopharmaceutic data). Please contact streamlinedsubmission@tga.gov.au at the TGA for specific advice.
Conditions for using an overseas reference product
If you intend to use an overseas reference product, you must ensure it meets both the following conditions:
- registered in, and obtained from, a country or jurisdiction with a regulatory system comparable to Australia
- marketed in the country of origin by the same company as the Australian reference product, or by a corporate entity that has a licensing arrangement with the company.
If the reference product does not meet the conditions listed above, you must submit a bioequivalence study using the Australian reference product.
Demonstrating the overseas and Australian reference products are identical
You need to provide evidence that the overseas reference product is identical to the Australian reference product.
The level of evidence required varies depending on several factors. Use the evidence requirement decision tree below to determine which scenario you meet and what evidence you are required to submit. You can also use the checklist table to help determine the evidence required for each scenario.
Evidence requirement decision tree
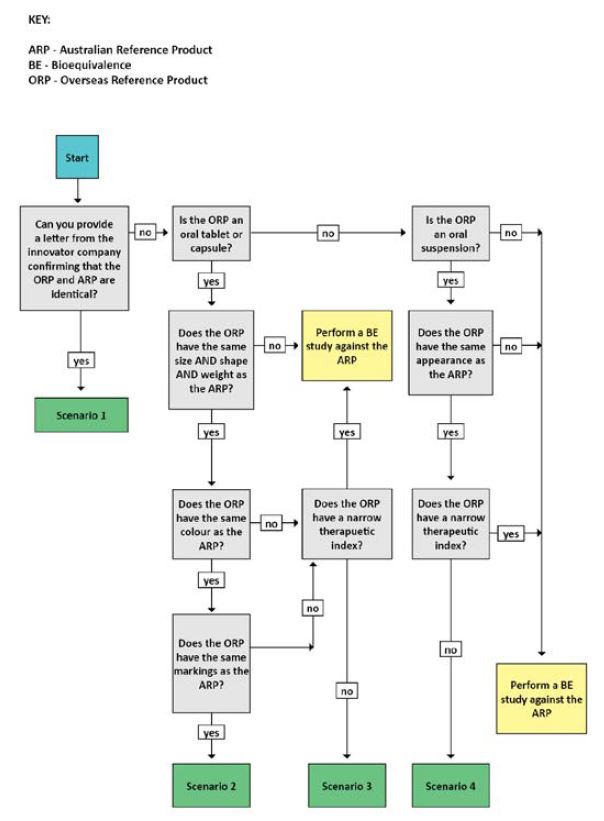
{kind=link}
Evidence requirement decision tree for biopharmaceutic studies.
Scenario 1
The following evidence is required:
- a declaration from the innovator company that the overseas and Australian reference products are identical in all respects including formulation and method of manufacture.
Scenario 2
The following evidence is required:
- evidence that the overseas and Australian reference products:
- have the same size, shape, weight, colour and markings
- contain the same nominal quantity of drug substance
- have the same qualitative formulation (e.g. as detailed in Product Information documents)
- copies of the labels and Product Information (or equivalent document) for both the overseas and Australian reference products
- certificates of analysis for both the overseas and Australian reference products analysed using the specifications proposed in the application for the generic medicine
- comparative dissolution profiles (see Comparative dissolution profiles for biopharmaceutic studies) of the overseas and the Australian reference products. The profiles should be determined across the physiological pH range (pH 1.2-6.8), including the Quality Control method.
For Scenario 2, the same evidence is required for immediate and modified release tablets and capsules.
Scenario 3
The following evidence is required:
- Evidence that the overseas and Australian reference products:
- have the same size, shape, weight
- do not have a narrow therapeutic index
- contain the same nominal quantity of drug substance
- have the same qualitative formulation, other than differences attributable to colours and/or markings (e.g. as detailed in Product Information documents).
- Copies of the labels and Product Information (or equivalent document) for both the overseas and Australian reference products.
- Certificates of analysis for both the overseas and Australian reference products analysed using the specifications proposed in the application for the generic medicine.
- Comparative dissolution profiles (see Comparative dissolution profiles for biopharmaceutic studies) of the overseas and the Australian reference products. The profiles should be determined across the physiological pH range (pH 1.2-6.8), including the Quality Control method.
- Evidence that the drug substance has a well-described dose-response curve and does not exhibit:
- a steep dose-response relationship
- a risk of serious undesired effects
- complicated or variable pharmacokinetics, such as:
- nonlinear pharmacokinetics
- variable or incomplete absorption
- an absorption window (i.e. site-specific absorption) or substantial (> 40 per cent) first-pass metabolism.
- Physicochemical evidence generated by an accredited laboratory demonstrating the overseas and Australian reference products are quantitatively identical.
- This may include Fourier transform infrared spectra, X-ray diffraction spectra, and full or partial quantitative chemical analyses (carried out in duplicate) of the excipients in those products.
- If a tablet is coated, provide spectroscopic and chemical analytical data for both the core and the coating, wherever possible.
- Provide data for at least two batches of each of the Australian and overseas reference products.
The precision and accuracy of the analytical methods and the inter-batch variability are critical to determining if the formulations are identical. The analytical methods and analytical method validation reports used to generate the physicochemical data are typically provided to satisfy this requirement.
As part of the validation of the test methods used, three batches of the proposed product should be tested with the test methods to demonstrate that the results obtained are both accurate and precise.
For modified release products:
Evidence is required to demonstrate that the Australian and overseas reference products have the same method of manufacture.
Scenario 4
The following evidence is required:
- Evidence to demonstrate that the overseas and Australian reference products:
- have the same appearance
- do not have a narrow therapeutic index
- contain the same nominal quantity of drug substance
- have the same qualitative formulation (e.g. as detailed in Product Information documents).
- Copies of the labels and Product Information (or equivalent document) for both the overseas and Australian reference products.
- Certificates of analysis for both the overseas and Australian reference products analysed using the specifications proposed in the application for the generic medicine.
- Comparative dissolution profiles (see Comparative dissolution profiles for biopharmaceutic studies) of the overseas and the Australian reference products. The profiles should be determined across the physiological pH range (pH 1.2-6.8), including the Quality Control method.
- Evidence that the overseas reference product has the same particle size distribution of suspended drug substance as the Australian reference product.
- Evidence that the overseas reference product has comparable re-suspension times to the Australian reference product.
- Physicochemical evidence. This might include, but not be limited to pH, buffer capacity, viscosity, surface tension and refractive index. Seek advice on relevant guidelines prior to submission if required.
Checklist table
The checklist table below may be used as a checklist for the evidence required for each scenario.
Grey boxes indicate that evidence is not required for the scenario. Further detail regarding these requirements can be found in the relevant scenarios detailed above.
| Evidence required | Scenario 1 | Scenario 2 | Scenario 3 | Scenario 4 |
|---|---|---|---|---|
| Declaration from the innovator company | Yes | No | No | No |
| Dosage form size, shape, weight, colour and markings | No | Yes | No | No |
| Dosage form size, shape and weight | No | Yes | No | No |
| Quantity of drug substance | No | Yes | Yes | Yes |
| Qualitative formulation | No | Yes | Yes | Yes |
| Labels and PI | No | Yes | Yes | Yes |
| Certificate of analysis | No | Yes | Yes | Yes |
| Dissolution profile | No | Yes | Yes | Yes |
| Drug substance is not narrow therapeutic index | No | No | Yes | Yes |
| The drug substance has a well-described dose-response curve | No | No | Yes | Yes |
| Physicochemical evidence | No | No | Yes | Yes |
| Method of manufacture (modified release products only) | No | No | Yes | No |
| Physical appearance | No | No | No | Yes |
| Particle size distribution of suspended drug substance | No | No | No | Yes |
| Re-suspension times | No | No | No | Yes |
Managing dropouts in bioequivalence studies
In a crossover bioequivalence study, some subjects will drop out of the study after (or even before) administration of the first treatment.
Manage dropouts by dosing several more than the required number of subjects in the first phase and specifying in the protocol how the requisite number of subjects will be chosen from those remaining in the study, for dosing in the second phase.
Related information and guidance
The FDA Guidance document: Statistical approaches to establishing bioequivalence.
Where to include biopharmaceutic data
Include: Biopharmaceutic studies in Module 5.3.1 of the CTD.
When submitting biopharmaceutic data:
The optional Bioequivalence Study Information Form may replace the requirement to complete the Summary of a Bioavailability or Bioequivalence Study form and some other requirements.
To find out if you can use this form, see guidance on Completing the Bioequivalence Study Information Form (BSIF).
- Complete the Summary of a Bioavailability or Bioequivalence Study form for each study.
- Include these forms in Module 1.11 of the CTD.
- Provide electronic and paper copies of the individual subject concentration versus time results, and the individual subject pharmacokinetic parameters from the pivotal biopharmaceutic studies.
- Include the study report and confirmation that all biopharmaceutic studies submitted to the TGA have been:
- carried out in accordance with the Declaration of Helsinki, and the principles of good clinical practice
- approved by an appropriate independent ethics committee or institutional review board.
- Identify and provide reasons, in Clinical Overview in Module 2 and Module 5.3.1 of the CTD, for any studies that have not been conducted in accordance with these requirements.
Justification for not submitting biopharmaceutic data
If biopharmaceutic data would normally be required but was not generated:
- include a justification for not submitting the data included in Module 1.9.2 of the CTD.
In preparing a justification:
Address at least the following issues, as applicable:
- the nature of the dosage form
- the solubility of the drug substance(s)
- the similarities of, or differences between, the formulations being considered
- the comparative dissolution profiles across the physiological pH range (1-7.5) of the products being considered
- the pharmacokinetic characteristics of the drug substance(s), such as permeability (or absolute bioavailability), linearity, first-pass effect (if any) and its significance
- the clinical consequences of any potential differences in bioavailabilities of the products under consideration (e.g. increased dose leading to toxicity or decreased dose leading to lack of efficacy)
- the margin between the minimum effective and minimum toxic plasma concentration.
Provide copies of any cited literature.
Use, where relevant, the Biopharmaceutic Classification System (BCS) to justify not undertaking in vivo bioequivalence studies. See ICH M9 guideline on biopharmaceutics classification system-based biowaivers.
The optional Biowaiver templates (BCS-based or additional strength) may replace the requirement to complete Module 2.5 of the CTD and some other requirements.
To find out if you can use these templates, see guidance on Completing the Biowaiver templates.
We do not have a list of drug substances that are considered to fall within particular BCS classes.
Page history
Title changed from 'Biopharmaceutic studies' to 'Providing biopharmaceutic data for medicine applications ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Description of changes (v1.2)
Title changed. Added links to new forms. Reduced requirements for use of overseas reference products in bioequivalence studies (no longer need to provide quantitative analysis of excipients for most solid oral dosage forms), Scientific Evaluation Branch
Description of changes (v1.1)
Minor text update, Scientific Evaluation and Special Product Access Branch
Description of changes (v1.0)
Original Publication, Office of Medicines Authorisation
Title changed from 'Biopharmaceutic studies' to 'Providing biopharmaceutic data for medicine applications ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Description of changes (v1.2)
Title changed. Added links to new forms. Reduced requirements for use of overseas reference products in bioequivalence studies (no longer need to provide quantitative analysis of excipients for most solid oral dosage forms), Scientific Evaluation Branch
Description of changes (v1.1)
Minor text update, Scientific Evaluation and Special Product Access Branch
Description of changes (v1.0)
Original Publication, Office of Medicines Authorisation