Pharmacovigilance Inspection Program metrics report: September 2017 to December 2018
Summary
As part of its Pharmacovigilance Inspection Program (PVIP), we conducted a total of ten inspections of Australian sponsors from 1 September 2017 to 31 December 2018.
A total of ten routine pharmacovigilance inspections were completed. There were no 'for-cause' inspections or re-inspections.
Inspections identified:
- no critical deficiencies
- 50 major deficiencies
- 29 minor deficiencies.
The common areas of findings are identified in Table 1.
All identified deficiencies have either been rectified or have Corrective and Preventative Actions (CAPAs) in place and are in the process of being resolved.
| Areas of findings | Critical (0) | Major (50) | Minor (29) |
|---|---|---|---|
| Management of significant safety issues | - | 9 | - |
| Collection and collation of adverse drug reactions | - | 12 | 3 |
| Management of adverse drug reactions | - | 9 | - |
| Ongoing safety evaluation | - | 9 | 5 |
| Management of reference safety information | - | 10 | 1 |
| Risk management | - | 1 | 3 |
| Australian pharmacovigilance contact person and the qualified person responsible for pharmacovigilance in Australia | - | 1 | - |
| Record-keeping requirement | - | - | 1 |
| Quality management system | - | 1 | 11 |
| Other post-approval commitments | - | 2 | 5 |
Table outlines the types of findings (and how critical they were) identified during pharmacovigilance inspections during the reporting period.
Background
Following a successful pilot program, the TGA launched the PVIP on 1 September 2017. The PVIP aims to strengthen and broaden the TGA's post-market monitoring activities and protect public health by ensuring the continued safety of medicines included on the Australian Register of Therapeutic Goods (ARTG).
Pharmacovigilance inspections allow the TGA to help sponsors meet their pharmacovigilance obligations and maintain effective and robust pharmacovigilance systems. The inspections assess the sponsor's compliance with currently applicable Australian pharmacovigilance regulations and guidelines, in particular:
- Therapeutic Goods Act 1989 (section 28(5e), 28(5)(ca), 28(2B), 28(3), 29A and 29AA)
- Therapeutic Goods Regulations 1990 (Regulation 15A)
- Pharmacovigilance responsibilities of medicine sponsors Australian recommendations and requirements (v2.1, June 2018)
- Conditions - standard and specific applying to registered or listed therapeutic goods (section 28 of the Act).
The TGA applies a risk-based approach to scheduling pharmacovigilance inspections. This takes into account risk factors relating to the sponsor, their products, their pharmacovigilance system and their compliance history. Additionally, the scores from the biennial PVIP Risk Assessment Survey are reviewed and considered in the inspection planning.
This first metrics report covers the 16-month period from PVIP commencement on 1 September 2017 to 31 December 2018, with the first inspection taking place in January 2018. The TGA intends to publish annual reports hereafter. The purpose of this report is to provide a high-level overview of inspection outcomes and common deficiencies to assist sponsors in improving their pharmacovigilance systems and in preparing for pharmacovigilance inspections. In the future, the report will also be able to show compliance trends and comparisons over time. All information has been de-identified.
Inspections conducted
From the commencement of the PVIP on 1 September 2017 to 31 December 2018, the TGA has conducted ten pharmacovigilance inspections of Australian medicine sponsors. The first inspection under the PVIP was undertaken in January 2018. All ten inspections were routine inspections, selected as a result of the TGA's risk-based scheduling process. Definitions of the inspection types are included in Appendix I.
A variety of sponsors were inspected during this period, including three sponsors who had products that included over-the-counter medicines, one of which also had herbal/complementary medicines. The remaining seven had prescription only medicines. In addition, three of the ten sponsors inspected were principally biosimilar and/or generic companies.
Findings identified during inspections were graded as critical, major or minor. The definitions for these are included in Appendix II. For the ten pharmacovigilance inspections, we found a total of:
- no critical findings
- 50 major findings
- 29 minor findings
These findings will be further discussed in this report.
Inspection findings
In this section: Critical findings | Major findings | Minor findings
Critical findings
No critical findings were identified in the ten inspections that were conducted during this period.
Major findings
There were 50 major findings identified across the ten inspections conducted during this reporting period. The number of major findings identified in each inspection ranged between four and six, as shown in the graph below.
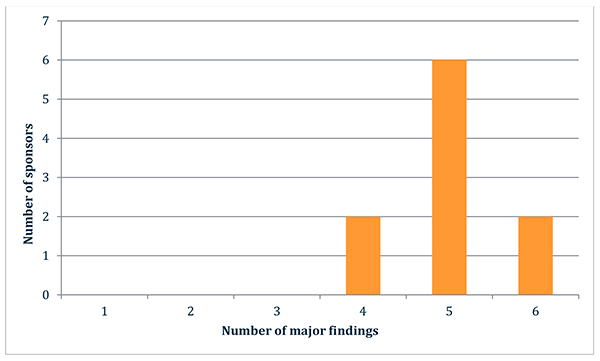
Figure is a graph showing the number of major findings identified during each pharmacovigilance inspection conducted during the reporting period.
Major findings by topic area
Findings have been grouped by overarching topics across the pharmacovigilance system, the nature of findings covered by each topic is provided in Appendix III.
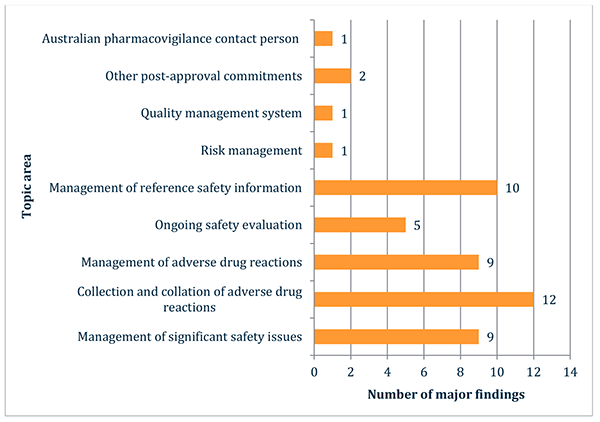
Figure is a graph showing the number of each type of major finding identified during the reporting period.
Summary of major findings reported during the period
The topic with the highest proportion of major findings in the reporting period was collection and collation of adverse drug reactions, followed by management of reference safety information, management of adverse drug reactions and management of significant safety issues. These common areas of major findings are detailed later in this report.
The major deficiencies identified have either been rectified or have CAPAs in place and are in the process of being resolved.
Minor findings
There were 29 minor findings identified across the ten inspections conducted during the reporting period. The number of minor findings identified in each inspection ranged between two and five, as shown in the graph below.
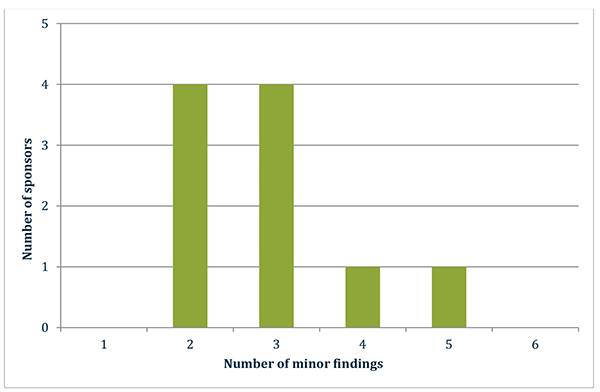
Figure is a graph showing the number of minor findings identified during each pharmacovigilance inspection conducted during the reporting period.
Minor findings by topic area
The chart below displays the distribution of minor findings by topic area:
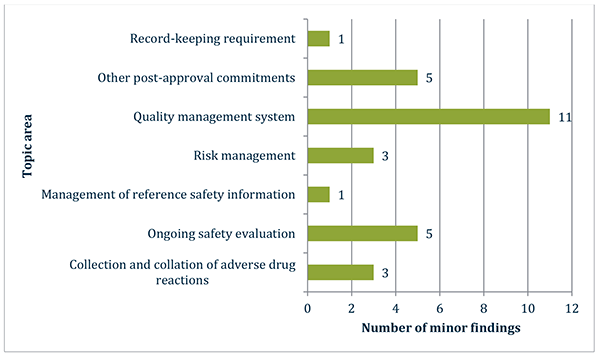
Figure is a graph showing the number of each type of minor finding identified during the reporting period.
Summary of minor findings reported during the period
The topic with the highest proportion of minor findings in the reporting period relates to deficiencies in the quality management system, in particular, deficiencies in procedural documents and pharmacovigilance training. This common area of minor findings is detailed in the next section of this report.
The minor deficiencies identified have either been rectified or have CAPAs in place and are in the process of being resolved.
Common areas of findings
In this section: Collection and collation of adverse drug reactions | Management of reference safety information | Management of adverse drug reactions | Management of significant safety issues | Quality management system
Collection and collation of adverse drug reactions
Deficiencies (any grading) related to the collection and collation of adverse drug reactions were identified in all ten inspections conducted during the reporting period. These include deficiencies in the:
- case collection from medical information enquiries, product quality complaints, company-sponsored websites and social media, patient support programs, registries and literature
- reconciliation of adverse reaction reports with internal departments, vendors and partners
- safety agreements and contracts between vendors and partners.
This topic also represented the highest proportion (24%) of all major findings.
Sponsors should have systems in place to identify, collect and collate all information related to the safety of their medicine - from all possible sources - in order to assess benefit-risk.
Sponsors should also exercise due diligence and have procedures in place to ensure adverse event reports are as accurate and complete as possible.
Management of reference safety information
Findings of any grading relating to the control and maintenance of reference safety information (RSI) were reported in all ten inspections conducted during the period. These included:
- delays in updating the Australian RSI after sponsor identification of the need for a safety-related change
- delays in updating the Australian Product Information (PI) and Consumer Medicine Information (CMI) documents outside the two weeks of the date of TGA approval as per the conditions of registration
- delays in updating the Australian PI and CMI of generics to align with the innovator outside the one month timeframe as per the specific conditions of registration
- delays in updating the minimum PI and package leaflets
- delays in the dissemination of updated Australian RSI to relevant company staff.
To ensure the safe use of medicines, it is necessary that all reference safety information documents available to health professionals, consumers and company personnel are kept up-to-date. RSI documents include but are not limited to PI and CMIs, as well as abridged PIs, promotional materials and medical enquiry standard responses.
Management of adverse drug reactions
Findings of any grading relating to the management of adverse drug reactions were reported in nine of the ten inspections conducted during the period. These included:
- non-submission or late submission of serious adverse reaction reports outside the regulatory 15 calendar days
- non-conservative seriousness assessment of adverse reaction reports
- inappropriate case processing of invalid reports which contain a drug-event pair
- inappropriate management of special situation reports such as off-label or pregnancy/breastfeeding cases
- deficiencies in the follow-up of adverse reaction reports, in particular case reports from literature and consumer reports.
Adverse reaction reports need to be appropriately assessed (for validation, seriousness, expectedness and causality), reported, coded and recorded into the company safety database to be used in ongoing monitoring. Sponsors should have procedures in place to regularly review the quality of case processing and to identify and address any process failures.
During this inspection period all observed delays in the reporting of serious adverse reaction cases by the sponsors were not considered to be system failures but occurred mostly due to individual case processing errors by company personnel.
Management of significant safety issues
Findings of any grading relating to the management of significant safety issues were reported in nine of the ten inspections conducted during the period. These were associated with failure to notify and/or late notification of significant safety issues outside the required 72 hour period.
A significant safety issue is a new safety issue or validated signal, considered by a sponsor in relation to their medicine. These issues require urgent notification to the TGA because of the seriousness and potential major impact on the benefit-risk balance of the medicine and/or on patient or public health, which could warrant prompt regulatory action and/or communication to patients and health professionals.
The TGA considers significant safety issues to include actions taken by comparable international regulatory agencies for safety reasons, such as the addition or modification of a contraindication, warning or precaution statement to the Product Information. The TGA expects sponsors to use clinical judgement when determining whether a safety issue is significant. Where after appropriate assessment the sponsor decides that a safety issue is not significant and hence not reportable, a justification should be documented.
Quality management system
Deficiencies (any grading) related to the quality management system were identified in all ten inspections conducted during the reporting period.
This topic represented the highest proportion (38%) of all minor findings and mostly concerned deficiencies related to procedural documents and pharmacovigilance training.
Sponsors should develop clear written Standard Operating Procedures (SOPs) for pharmacovigilance to ensure all roles, responsibilities, requirements and timelines are well-defined and understood by all personnel involved. Additionally, sponsors should provide appropriate training to all staff engaged in pharmacovigilance activities or who might receive or process safety reports. Level of training should reflect the employee's roles and responsibilities.
Comparisons of inspection findings over time
The TGA has inspected a total of 20 sponsors to date, including ten volunteer companies that took part in the pilot program undertaken in 2015-16. The details and conclusions from the TGA Pharmacovigilance Inspection Pilot Program can be found at: Presentation: The TGA Pharmacovigilance Inspection Pilot Program: 2015-2016.
Case collection and processing of adverse reaction reports and the management of reference safety information continue to be common areas for findings.
Appendix I: Inspection type definitions
*excerpt from page 10-11 of the Pharmacovigilance inspection program: Guidance for medicine sponsors (Version 1.0, September 2017). Please note the TGA is referred to as 'we' or 'us', and sponsors as 'you'.
Routine inspections
Routine pharmacovigilance inspections are scheduled as part of the inspection program. There is no specific trigger for these inspections, although we take a risk-based approach to prioritising them. These inspections are usually system-related inspections, but one or more products may be selected as examples to verify the implementation of the system and provide practical evidence of its functioning and compliance.
'For cause' inspections
'For cause' inspections are undertaken in response to specific triggers where a pharmacovigilance inspection is the appropriate way to examine the issues. 'For cause' inspections generally focus on specific aspects of the sponsor's pharmacovigilance system or examine identified compliance issues and their impact on a specific product. However, we may also inspect the sponsor's entire pharmacovigilance system as a result of a trigger. Significant public health concerns or identified noncompliance are expected to be the most common triggers.
Re-inspections
We may re-inspect the sponsor's pharmacovigilance system as part of our routine inspection program. We prioritise re-inspections by assessing risk factors. If a previous inspection identified a high level of compliance this may increase the time between re-inspections. More frequent re-inspections may occur:
- where we have identified significant noncompliance
- to verify sponsors have taken action to address inspection findings
- to evaluate the sponsor's ongoing compliance with their obligations and evaluate changes to their pharmacovigilance system
- when a previous inspection finds a sponsor had failed to take appropriate corrective and preventative action in response to prior inspections.
Appendix II: Inspection finding definitions
*excerpt from page 211 of the Pharmacovigilance inspection program: Guidance for medicine sponsors (Version 1.0, September 2017)
Critical deficiency:
A deficiency in pharmacovigilance systems, practices or processes that adversely affects the rights, safety or well-being of patients or that poses a potential risk to public health or that represents a serious violation of applicable legislation and guidelines.
Deficiencies classified as critical may include a pattern of deviations classified as major.
A critical deficiency also occurs when a sponsor is observed to have engaged in fraud, misrepresentation or falsification of data.
Major deficiency:
A deficiency in pharmacovigilance systems, practices or processes that could potentially adversely affect the rights, safety or well-being of patients or that could potentially pose a risk to public health or that represents a violation of applicable legislation and guidelines.
Deficiencies classified as major may include a pattern of deviations classified as minor.
Minor deficiency:
A deficiency in pharmacovigilance systems, practices or processes that would not be expected to adversely affect the rights, safety or well-being of patients.
A deficiency may be minor either because it is judged as minor or because there is insufficient information to classify it as major or critical.
Deficiencies are classified by the assessed risk level and may vary depending on the nature of medicine. In some circumstances an otherwise major deficiency may be categorised as critical. A deficiency reported after a previous inspection and not corrected may be given higher classification.
Appendix III: Categorisation of findings
| Topic area | Sub-topic of reported findings |
|---|---|
| Management of significant safety issues | Assessment and notification of significant safety issues |
| Collection and collation of adverse drug reactions | Case collection, including monitoring, identification and reconciliation |
| Spontaneous sources of safety data e.g. medical information, product quality complaints | |
| Literature searching | |
| Solicited sources of safety data, including patient support or market research programs | |
| Safety data exchange agreements | |
| Management of adverse drug reactions | Case processing, including data entry, coding, causality and seriousness assessment, follow-up and reporting |
| Data management, including migration of safety data | |
| Ongoing safety evaluation | Ongoing monitoring, signal detection |
| Periodic safety update reports | |
| Management of reference safety information | Maintenance of reference safety information, including the Company Core Data Sheets, Product Information (PI) and Consumer Medicine Information (CMI), minimum PI and package leaflets |
| Maintenance of safety information on sponsor websites | |
| Dissemination and communication of Australian Reference Safety Information (internally and externally) | |
| Risk management | Management of additional pharmacovigilance activities in the Risk Management Plan (RMP), for example Post-Authorisation Safety Study, targeted follow-up questionnaires |
| Management of additional risk minimisation activities in the RMP | |
| Safety communication, for example Dear Health Care Professional Letter | |
| RMP and Australian‐Specific Annex maintenance | |
| Australian pharmacovigilance contact person and the qualified person responsible for pharmacovigilance in Australia (QPPVA) | Notification of the Australian pharmacovigilance contact person |
| Pharmacovigilance system oversight and governance, including performance monitoring and role of the QPPVA | |
| Record-keeping requirement | Retention of pharmacovigilance records |
| Quality management system | Procedures, record management, pharmacovigilance training |
| Audit and deviation management, including Corrective and Preventive Action management | |
| Information technology systems and applications | |
| Other post-approval commitments | For example specific conditions of registration, advertising code of conduct |
Table outlines the topic areas and sub-topics used to categorise findings of pharmacovigilance inspections during the reporting period.