Application instructions for Conformity Assessment certification
How to apply for a Conformity Assessment certificate.
Read this in conjunction with the Australian Regulatory Guidelines for Medical Devices (ARGMD)
TGA identifiers
We use a several identifiers to track elements of the conformity assessment application process.
These are:
| Identifier | Format and example | Description and use |
|---|---|---|
| Submission ID | Format: DC-YYYY-xxxx-1 Example: DC-2022-54321-1 | The primary ID used by the TGA to identify the application during the assessment process Should be included on all correspondence with the TGA Sometimes referred to as the 'DC' number Generated after the application fees are paid |
| Application Identifier | Format: DV-YYYY-CA-xxxxx-1 Example: DV-2022-CA-54321-1 | Used to identify the electronic application Rarely referred to during a conformity assessment certification application Generated when a draft application is first saved |
Record Number or File Number | Format: YYYY/xxxxxx Example: 2022/001234 | This tracks the file(s) associated with an application or manufacturer. All correspondence with a manufacturer and all TGA reports are placed on the file (or series of files) or associated data container. |
| Certificate Number | Format:
Examples:
Format:
Examples:
| This identifier identifies a manufacturer's certificate(s).
|
| Version Number | Format: R.C Example: 2.1 | What we use to identify the version of the certificate
|
| Manufacturer Root File Number | Format: YYYY/xxxxxx Example: 2021/001234 | The identifier for the file used within the TGA to store the manufacturer's certificates. |
How to apply
Step 1: Create an e-Business account
If you do not have an account, see
TGA business services: getting started with the TGA
The e-Business forms are at TGA business-services-forms on the TGA website.
Step 2: Lodge an electronic application
Submit your applications for conformity assessment through TGA Business Services.
- Select Open TGA Business Services
- Login to enter the system.

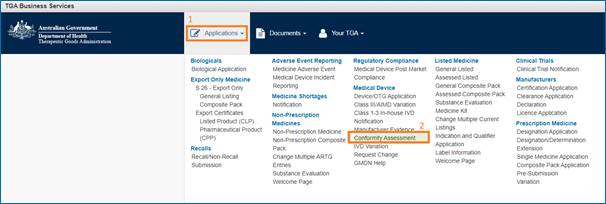
Once you've access to the eBS system,lodge an electronic application.
- Click on Applications (1)
- Select Medical Devices: Conformity Assessment (2)
- Complete the application form.
Initial applications
When making an initial application for a new certificate (for example, for an initial assessment of the manufacturer's quality management system or for a new kind of device), the applicant should include a description of the new entity / device in the applicant's reference field. If possible, also indicate if a subsequent change to an associated certificate is required.
For example:

Electronic attachments should not be included when submitting the electronic application form for a Conformity Assessment Certificate. Supporting information is submitted separately to us (see below) only on request.
Substantial change notification and application
When making a substantial change notification and application the applicant should write 'Change to [aspect] for certificate(s): [Certificate numbers]' in the Applicant's Reference field. For example:

Recertification applications
When making an application for recertification of an expiring certificate, the applicant should write "Recertification for certificate(s): [Certificate IDs separated by commas]" in the applicant's reference field.
For example:

Step 3: Payment of application fee
Upon submission of the electronic application form, an application fee is payable to us. Payment is made through the online invoice payment portal included in the eBS portal.
Failure to pay the application fee will result in the application being ineffective and consequently it will automatically lapse.

You will find the application fee under 'Application Type Details'.

Information on applications fees can be found on the fees and charges page.
Applicable assessment and onsite inspection fees are set at a later stage and are invoiced separately.
Step 4: Supporting documentation
Once the electronic application is submitted and the application fee is paid, the application will be assigned a Submission ID (format DC-YYYY-xxxx-1). All documents and correspondence regarding the application should reference the assigned Submission ID.
We will send the applicant an acknowledgement email and request for supporting information. This request may require the applicant to complete a form "Supporting Data Form—Conformity Assessment Certification" and to provide supporting data. The form and associated data should only be provided when requested.
The form is available under forms at Application for conformity assessment certificates (medical devices).
If you are a manufacturer applying for a Conformity Assessment Certificate, you should have in place a quality management system and technical documentation that complies with the requirements of the Therapeutic Goods (Medical Devices) Regulations 2002 (The Regulations). If this is not the case, you should apply at a later stage when they are ready. Submission of insufficient supporting data may lead to applications being lapsed.
Formatting instructions for supporting data
Information and data should be organised for ease of reference (for example, in a folder hierarchy, bookmarks in PDF document), and a table of contents provided.
Cover letter
You will need to provide a cover letter.
Any information that's not in the supporting data form should be in the cover letter.
If there are similarities between the device you are applying for and the predicate device, produced by the same manufacturer that is supported by TGA conformity assessment certificate, you are advised (in tabular format within in the cover letter) to clearly highlight the similarities and differences between the new device and the predicate device with respect to their design, construction, materials (including formulation), intended purpose, administration, packaging materials, sterilisation process and shelf-life.
Supporting documents
All supporting data and documents must be provided in a readable electronic format:
- Data and documents should be provided in PDF format as individual files (i.e. not joined together in a single PDF file). Other common file types (such as RTF or MS Word) may be accepted if PDF format is unavailable.
- The data provided must be searchable (either by generating the documents electronically and converting to PDF format, or by scanning in documents using optical character recognition (OCR)).
- Initial supporting data should be provided on a CD or DVD (preferably non-rewritable CD-R or DVD-R).
- For data files exceeding 15mb, submission via a temporary online portal is the preferred method. Contact the medical devices team at devices@tga.gov.au to request access.
- Due to archiving and storage requirements, data submitted on USB storage media (or other electronic media such as hard drives) may not be accepted.
We will accept submissions in the IMDRF Non-In Vitro Diagnostic Device Market Authorization Table of Contents and the In Vitro Diagnostic Medical Device Market Authorization Table of Contents format provided that the document contains the requested information.
See the IMDRF website at IMDRF website for more.
Posting supporting documentation
When requested to provide documentation, forward your response to:
| Postal address | Courier delivery | |
|---|---|---|
| Devices Application and Triage Section Medical Devices Authorisation Branch Therapeutic Goods Administration PO Box 100 WODEN ACT 2606 | or | Devices Application and Triage Section Medical Devices Authorisation Branch Therapeutic Goods Administration 27 Scherger Drive FAIRBAIRN ACT 2609 |
If you have any questions, call the Medical Devices Information Line on 1800 141 144.
Next steps
As soon as we get the application and supporting documents, we can start the pre-assessment.
This is a preliminary screening of the provided supporting documentation.
If we find that in general the applicant has not provided sufficient information to allow commencement of assessment, the application may lapse.
Further information for clarification purposes may be necessary at the pre-assessment stage.
We may send a request for information to the applicant under section 41JA of the Therapeutic Goods Act 1989 (the Act).
When this happens, the assessment clock for the application is stopped. The assessment clock restarts when we receive all responses back.
Failure to provide the required information and/or data within the time specified in the request for information will result in the application automatically lapsing.
If the application lapses, the applicant will need to make a new application.
The pre-assessment results in the creation of an Assessment Plan and a determination of the associated assessment fees required to undertake assessment for the relevant application.
Extension of time
Extensions for applicants to respond to s41JA requests may be granted. This is only in extenuating circumstances.
Circumstances might be:
- manufacturer severely affected by a natural disaster (e.g., severe flood, earthquake, fire)
- death or serious illness of key personnel within the manufacturer's facility(ies) normally responsible for obtaining such information
- manufacturer's premises recently destroyed by fire or damage/loss as a result of theft or break-in
Situations that would not be extenuating include:
- the manufacturer being away on holidays
- you have only checked your mail or have not realised that we had requested the information
- the manufacturer needing time to have the information translated
- the manufacturer needing extra time to generate the documents or conduct testing
You must make requests for extensions of time as early as possible (and before the due date). Include the reason for the extension, as well as a suggested timeframe.
Extensions of time may be made to the delegate requesting the information.
Requests for extensions are considered on a case-by-case basis. An extension shouldn't be taken as a precedent for future requests.
Acceptance and assessment fees and payment
Once your submission is accepted for assessment, we'll email you about fees.
For more information, see Fees and payments. Also see, Reduction of assessment fees for medical devices.
Assessment
Following receipt of the assessment fees, the supporting data are sent to the relevant technical teams for assessment.
Further information for clarification purposes may be necessary during the assessment stage. The TGA may send a request for information to the applicant under section 41JA of the Therapeutic Goods Act 1989 (the Act). Failure to provide the required information and/or data within the time specified in the request for information will result in the application automatically lapsing. If the application lapses, the applicant will need to make a new application.
Quality management system audits
If an audit of the manufacturer is required, this will be scheduled by the Devices Quality Audits and Assessments Section (DQAAS) of the TGA. An officer from DQAAS will contact the manufacturer to arrange a suitable audit time. Additional audit fees such as any travel-associated expenses for the audit and any audit time in excess of two audit days will be invoiced separately.
Type examination
If the application is for Type Examination certificate requiring testing, analysis, and reporting on examination of the type as described in Schedule 3, Part 2 of the Therapeutic Goods (Medical Devices) Regulations 2002, an additional fee to cover the costs associated with testing the relevant kind of medical device is also payable. The fee is the amount that reimburses the Department for the costs incurred in purchasing, establishing and setting‑up the equipment to be used to conduct the tests and the direct costs of conducting the tests (including the cost of any consumables used in conducting the tests).
Supplementary assessment
If the application is for a medical device that requires supplementary assessment (for example for devices containing a medicinal substance) additional assessment fees prescribed in Item 1.12 of Schedule 5 of the Therapeutic Goods (Medical Devices) Regulations 2002 may be applicable. If the medicinal substance contained in the medical device is a new chemical entity, additional fees prescribed under Item 4, or paragraph (b) or (d) of Item 5, of Part 2 of Schedule 9 to the Therapeutic Goods Regulations 1990- external site may be applicable for assessment of the data relating to this medicinal substance.
Timeframes
Information on conformity assessment timeframes can be found within the Conformity Assessment process and timeframes page of our website.
Advisory Committee on Medical Devices (ACMD)
Applications for high-risk devices, or with novel technology, may involve consultation with the Advisory Committee on Medical Devices (ACMD).
The ACMD is a panel of academic and clinical specialist experts. It also includes a consumer representative.
In these cases, our assessors pose specific questions that are relevant to the assessment of the application, to the committee.
If we decide to seek advice from the ACMD, we will let you know before the meeting.
Following the meeting, the applicant will be provided the questions posed to the ACMD and the responses provided by the committee.
Applicant's Statutory Declaration
The Secretary, or delegate, must consider whether an applicant has during the period of 10 years immediately before the application failed to meet one or more of a number of specified criteria.
See Manufacturer Statutory Declarations.
The applicant's declaration is requested as part of the initial supporting data. To complete the form go to Manufacturer Statutory Declarations and select certificate S41EC(3)(a).
To reduce processing times, send the completed applicant's declaration by email to devices@tga.gov.au.
Make sure your email identifies the submission identification number (i.e., Submission ID) of the associated Conformity Assessment Application.
Issuing of certificates
Certificates will only be issued if the following conditions are met:
- the assessment of the device's compliance to the Essential Principles is completed and demonstrated by the manufacturer;
- the quality management system audit (if conducted) is closed out - that is, all non-conformities are resolved;
- all contractual arrangements for CE Marking (if applicable) are completed;
- all clearances (including the certificate under section 41EC, if applicable) are completed; and
- all fees (application, assessment, additional audit fees, etc.) are paid in full.
Certificates are issued to the manufacturer by the Devices Conformity Assessment Section of the Medical Devices Authorisation Branch (MDAB). Sponsors can use the certificate(s) as evidence to support inclusion of the medical device(s) in the Australian Register of Therapeutic Goods (ARTG) in order for its lawful supply in the Australian market.
Legal supply in Australia
Once a certificate is issued to a manufacturer, sponsors can:
- lodge the new certificate as Manufacturer's Evidence (ME); and
- apply for inclusion of the device(s) in the ARTG
A medical device cannot be lawfully supplied in Australia unless the Australian Sponsor holds a current ARTG entry for the device. Instructions for submitting the Manufacturer's Evidence and for making applications for medical device inclusion are provided to applicants when the certificates are issued.
Note:
- Applications for ME take up to 15 TGA working days to process.
- Applications for inclusion of medical devices in the ARTG may be made upon acceptance of the ME and can take up to 5 TGA working days to process when TGA certificates are used as ME.
- ME applications supported by TGA Conformity Assessment Certificates and applications for inclusion of the device(s) in the ARTG are processed by the Devices Application Section of MDAB.
- Should certificates be reissued by the delegate (for example, following a substantial change notification or recertification application), Australian sponsors may need to submit the new certificate as a variation to their ME. Depending on the type of change, sponsors may also need to make a new medical device inclusion application or a Device Change Request application through the sponsor portal.
Substantial change notification and application
A manufacturer holding a TGA conformity assessment certificate is subject to automatic conditions imposed under section 41EJ of the Act.
They are required to make sure they comply with applicable conformity assessment procedures.
One of these being to notify us of any plan for substantial changes to:
- quality management systems
- the product range covered by those systems
- the product design of kinds of medical devices
Substantial change notification and application can be made using the e-Business system.
Changes to any of the following constitute a 'substantial change'.
These include changes to:
- any detail on the certificate including manufacturer's name or address
- device category, or to unique product identifiers
- additions or deletions of new critical suppliers
- critical processes (for example, change of sterilisation method)
- manufacturer's facility name or address
- the design of the device
- source for materials of animal origin
- the formulation of the IVD reagent
- stability aspects of the IVD reagent (extension/updates to on-board stability)
- extension to specimen stability claims to include cadaveric specimen testing using infectious disease assays
- existing kind of device to include a new immunohaematology reagent (IHR)
Recertification of an expiring certificate
Recertification applications should be within six months of the manufacturer's certificate(s) expiry.
Manufacturers are responsible for updating contact info in the e-Business portal.
It is on them to make sure recertification applications are made in sufficient time for the application to be completed and new certificates to be issued.
Once new certificates are issued, Australian sponsors may need to submit the new certificate as a variation to ME.
Depending on the change, sponsors may need to:
- make a new medical device inclusion application or
- a Device Change Request application through the sponsor portal.
Application process flowchart
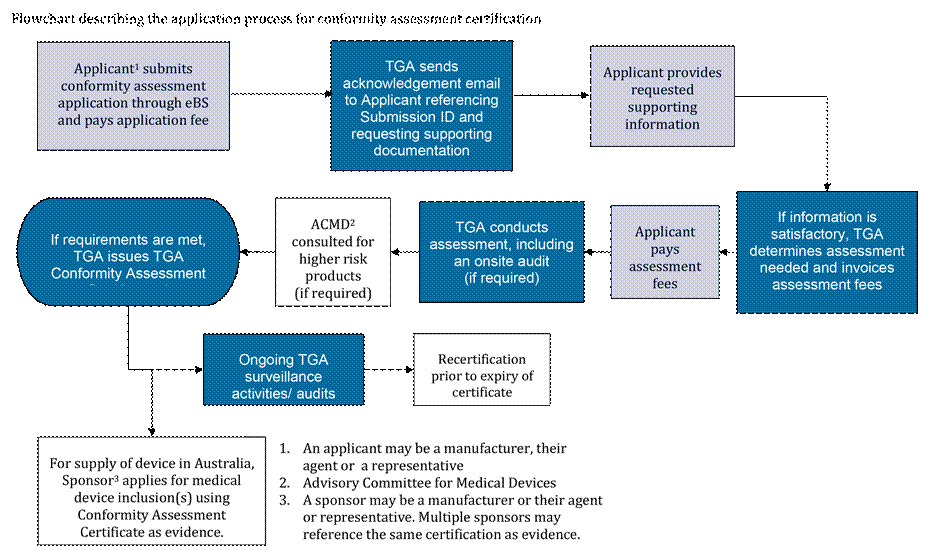
Text version of flowchart
This flowchart is represented as a list with numbered steps.
- Applicant submits conformity assessment application through eBS and pays application fee
- An acknowledgement email goes to Applicant referencing Submission ID and requesting supporting documentation
- Applicant provides requested supporting information
- If information is satisfactory, we determine assessment needed and invoices assessment fees
- Applicant pays assessment fees
- We conduct assessment, including an onsite inspection, if required
- ACMD consulted for higher risk products (if required)
- If requirements are met, we issue TGA Conformity Assessment Certificate(s)
- For supply of device in Australia, Sponsor applies for device inclusion(s) using Conformity Assessment Certificate as evidence - end flowchart
- Ongoing TGA surveillance activities/audits
- Recertification before expiry of certificate
- End flowchart
Page history
Redaction of outdated conformity assessment processing times and fees.
Updated medical device contact email address.
Updated Department logo, copyright information, links and eBS information.
Change of ACMD process and recertification submission timeframe.
Added:
- Manufacturer Root File Number to Reference: TGA Identifiers table
- Applications for extension of time to submit responses to s41JA requests
Change of heading
From Fit-and-Proper Person Certification to Considering Applications Under Section 41EC (Fit-and-Proper Person)
Redaction of outdated conformity assessment processing times and fees.
Updated medical device contact email address.
Updated Department logo, copyright information, links and eBS information.
Change of ACMD process and recertification submission timeframe.
Added:
- Manufacturer Root File Number to Reference: TGA Identifiers table
- Applications for extension of time to submit responses to s41JA requests
Change of heading
From Fit-and-Proper Person Certification to Considering Applications Under Section 41EC (Fit-and-Proper Person)