Website maintenance took place on Thursday 30 April 2026. If you experience any issues, please contact us.
Introduction
Pharmacovigilance is defined by the World Health Organization as the science and activities related to detecting, assessing, understanding and preventing adverse effects and other medicine-related problems. The Therapeutic Goods Administration (TGA) collects and evaluates information related to the benefit-risk balance of medicines in Australia to monitor their safety and, where necessary, take appropriate action.
This guidance sets out the pharmacovigilance responsibilities of sponsors of medicines included on the Australian Register of Therapeutic Goods (ARTG) and regulated by the TGA. It outlines the mandatory reporting requirements and offers recommendations on pharmacovigilance best practice.
In this guidance we use ‘must’ or ‘required’ to describe something you are legally obliged to do. We use ‘should’ to recommend an action that will assist you to meet your legal requirements. We refer to the TGA as ‘we’ or ‘us’, and to sponsors as ‘you’.
Scope
This guidance:
- applies to all sponsors who have medicines registered or listed on the ARTG
- describes your pharmacovigilance reporting and record-keeping requirements
- offers recommendations on the monitoring, collection and management of safety data to help you achieve best practice pharmacovigilance
- outlines the legal basis for pharmacovigilance requirements associated with medicines on the ARTG.
This guidance is mostly consistent with the European Medicines Agency’s EMA Guideline on Good Pharmacovigilance Practices (GVP) Module VI—Management and reporting of adverse reactions to medicines. Some requirements and recommendations, however, are specific to Australia.
Responsibilities
You must meet your pharmacovigilance reporting responsibilities for all the medicines you have registered or listed on the ARTG. This is regardless of their Australian marketing status—that is, whether they are currently available for purchase, withdrawn from sale or otherwise supplied (e.g. in a company-sponsored post-registration study).
Unapproved medicines used in clinical trials or supplied under the Special Access Scheme or Authorised Prescriber Scheme are subject to separate reporting requirements and are not covered by these guidelines.
You, as a sponsor of medicines approved for supply in Australia, are legally responsible for meeting pharmacovigilance reporting requirements for your medicine.
You must:
- let us know who your Australian pharmacovigilance contact person is
- submit any serious adverse reaction reports to us
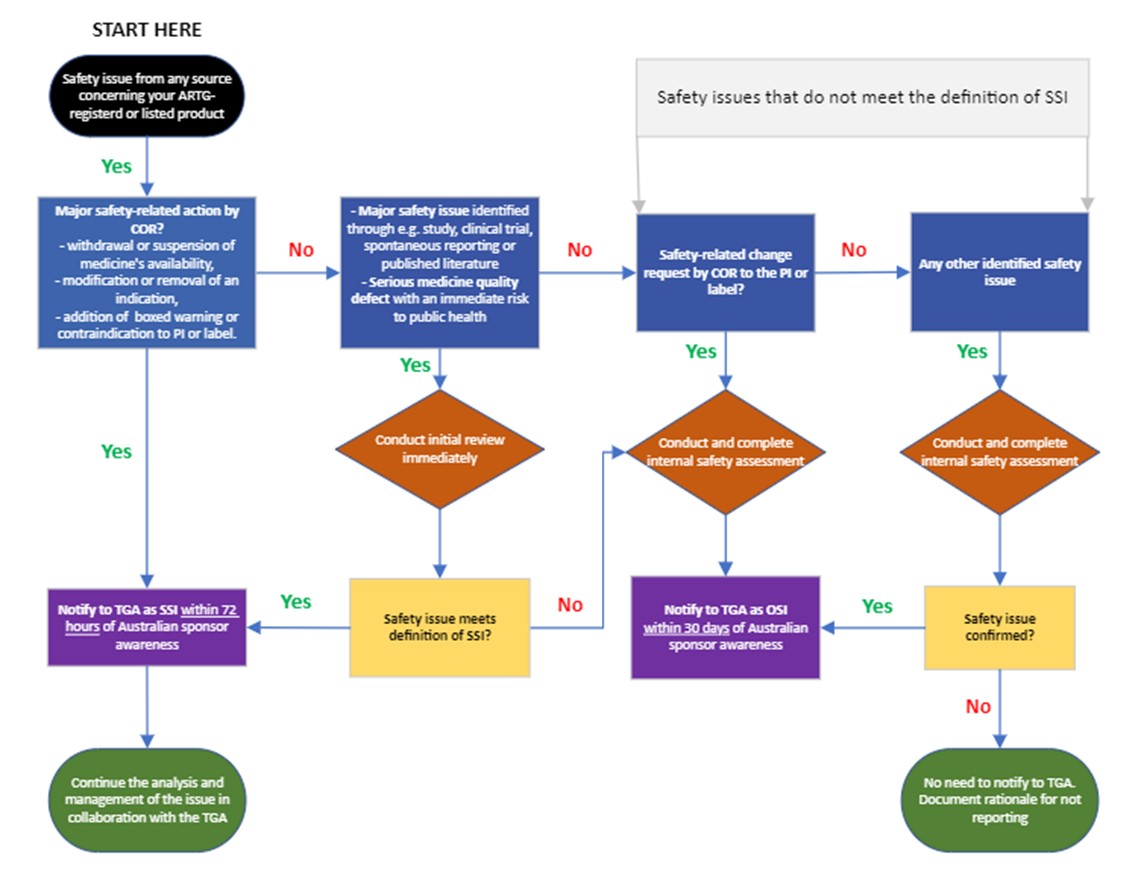
- notify us of any safety issues you identify
- keep records pertaining to the reporting requirements and safety for your medicine (under Subsection 28(5)(ca) of the Therapeutic Goods Act 1989 (the Act))
- answer any request from us for additional information fully and within the specified timeframe (under Subsection 31(1) of the Act).
This includes situations where:
- separate sponsors market two or more separately registered or listed medicines that are considered identical in all respects apart from their trade names
- sponsors share or outsource marketing or distribution arrangements
- pharmacovigilance activities are contracted to external organisations.
We require you to have an effective pharmacovigilance system in place in order to:
- collect and collate safety information pertaining to your medicine
- monitor and take responsibility for the safety of your medicine
- meet legislative requirements for reporting serious adverse reactions and safety issues
- identify any changes to the benefit-risk balance of your medicine
- take appropriate action in a timely manner when necessary
- update product labels and product information (PI) documents with new safety information in a timely way.
Related information and guidance
Questions relating to this guidance document may be addressed to Pharmacovigilance.Enquiries@health.gov.au
{kind=link}